Transferir como PDF, PPTX



























































![ENTALPIA DE REAÇÃO QUÍMICA
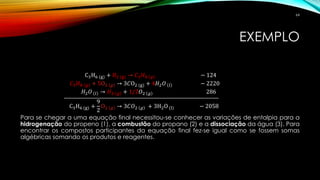
Analisa-se abaixo a reação de formação da água:
H2 (g) +
1
2
O2 (𝑔) → H2O (g)
Cálculo da variação de entalpia padrão:
∆Hr,m = 𝐻𝑓,𝑚 H2O, g − [𝐻𝑓,𝑚 H2, g +
1
2
𝐻𝑓,𝑚 O2, g ]
Percebe-se que o cálculo da variação de entalpia numa reação química é direto, mas deve-
se tem em conta todos os produtos somados entre si menos todos os reagentes somados entre
si. Esses valores calculados são de átomos puros isolados em estado padrão, então por isso os
valores de cada um são analisados individualmente e depois somados entre si.
Para uma equação qualquer tem-se:
aA + bB → cC + dD
∆Hr,m = 𝐻𝑓,𝑚cC + 𝐻𝑓,𝑚dD − 𝐻𝑓,𝑚aA + 𝐻𝑓,𝑚bB
60](https://image.slidesharecdn.com/introduosleisdatermodinmica-170530070540/85/Introducao-as-leis-da-termodinamica-60-320.jpg)

































![ENTROPIA PADRÃO DE REAÇÃO
Como agora a entropia de uma substância pode ser calculada as reações químicas agora
também poderão, do mesmo jeito que na entalpia de reação.
Analisa-se abaixo a reação de formação da água:
H2 (g) +
1
2
O2 (𝑔) → H2O (g)
Cálculo da variação de entalpia padrão:
∆Sr,m = 𝑆 𝑚 H2O, g − [𝑆 𝑚 H2, g +
1
2
𝑆 𝑚 O2, g ]
Para uma equação qualquer tem-se:
aA + bB → cC + dD
∆Sr,m = 𝑆 𝑚cC + 𝑆 𝑚dD − 𝑆 𝑚aA + 𝑆 𝑚bB
97](https://image.slidesharecdn.com/introduosleisdatermodinmica-170530070540/85/Introducao-as-leis-da-termodinamica-97-320.jpg)


1) O documento introduz os conceitos fundamentais da termodinâmica, incluindo sistema, vizinhança, propriedades extensivas e intensivas, e estado do sistema. 2) A primeira lei da termodinâmica é explicada, estabelecendo que a energia interna de um sistema pode ser alterada por meio do trabalho ou do calor. 3) A diferença entre trabalho realizado pelo sistema e trabalho realizado sobre o sistema é definida com exemplos.









































































