Baixado 22 vezes






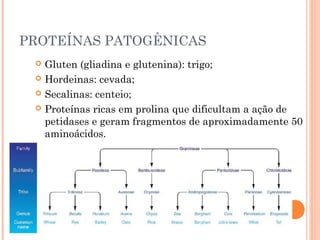

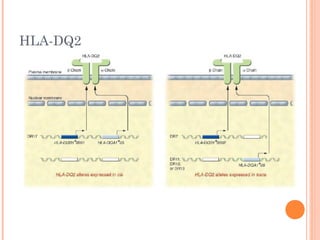




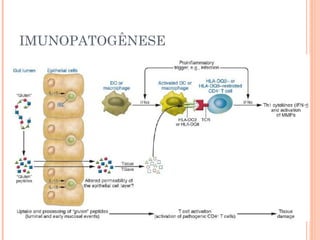

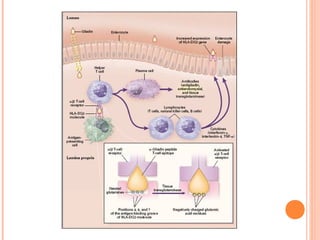
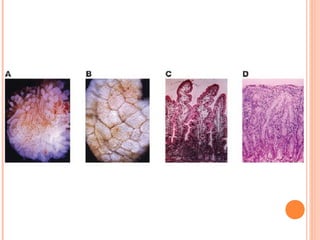












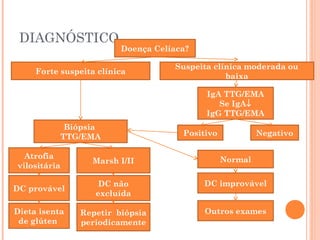
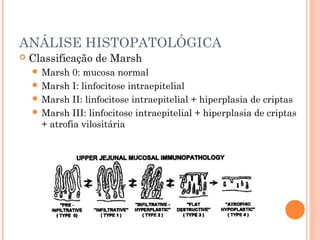
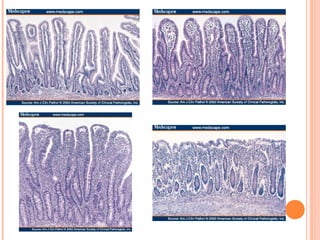






Este documento descreve a doença celíaca, uma condição autoimune causada pela ingestão de glúten que afeta o intestino delgado. Apresenta os sintomas típicos e atípicos, fatores de risco genéticos e ambientais, métodos de diagnóstico e o tratamento baseado na exclusão do glúten da dieta.















































