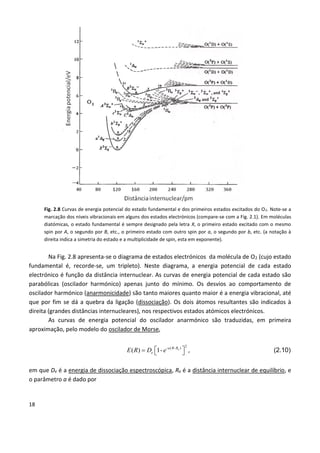
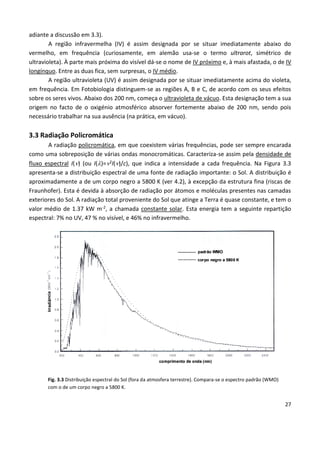
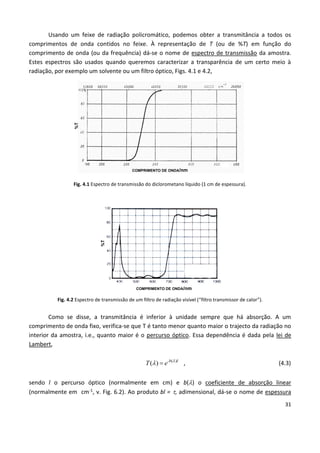
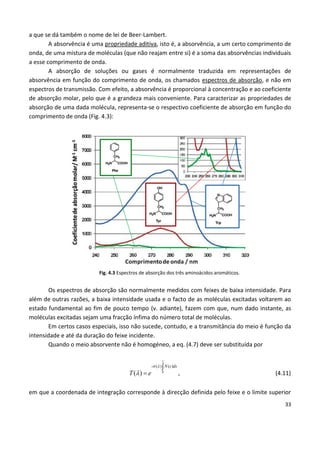
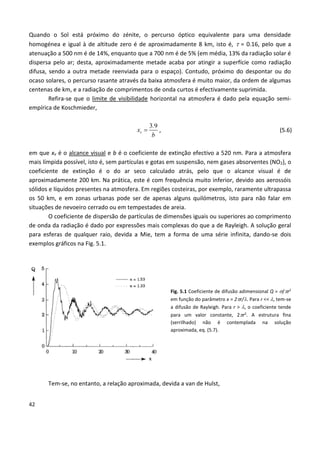
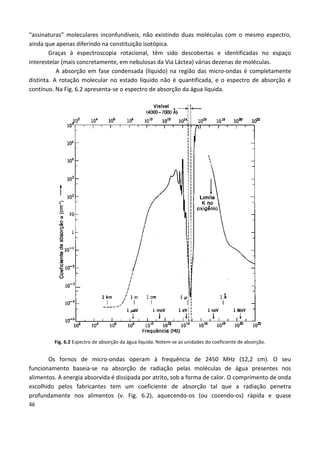
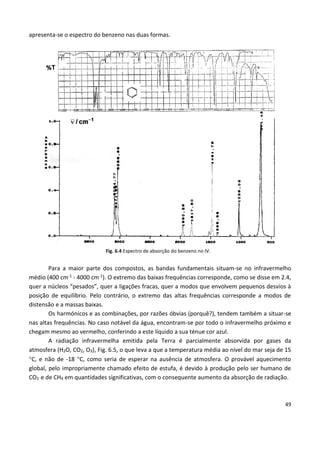
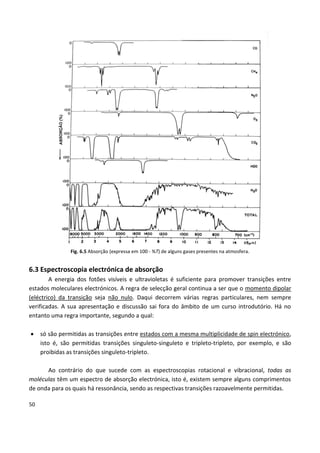
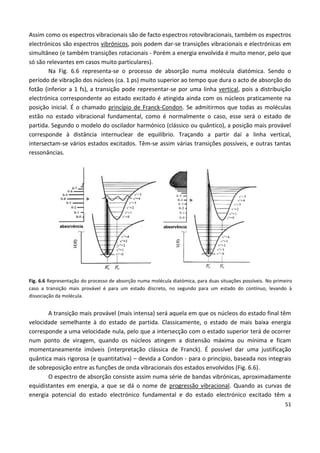
Este documento fornece uma introdução abrangente à espectroscopia. Resume os principais pontos da seguinte maneira:
1) A espectroscopia estuda a interação entre radiação e matéria, fornecendo informações sobre a estrutura, dinâmica e propriedades químicas e físicas de átomos, moléculas e outros sistemas.
2) O desenvolvimento da espectroscopia foi fundamental para o estabelecimento da teoria quântica e levou a várias descobertas químic






![6
esféricos, pois a propriedade de polarização da luz implicava alguma dissimetria. Em Ciência, muitas
vezes «o objectivo não é tanto ver o que ainda ninguém viu, mas sim pensar o que ninguém ainda
pensou, sobre aquilo que todos vêem» (Schrödinger)!
Fig. 1.4 Gravura feita a partir do desenho original de Newton
sobre a sua experiência. Nec variat lux fracta colorem, ou seja, e
mantendo a ordem das palavras - para acentuar a proximidade
entre português e latim - Não varia a luz fraccionada [refractada]
na cor. In A. Secchi, Le Soleil, 2ª ed., Paris, 1875.
O termo Espectroscopia começou a ser usado em finais do séc. XIX, depois de Bunsen e
Kirchhoff terem verificado, em 1860, que os espectros de absorção e de emissão permitiam
determinar a presença de certos elementos químicos numa amostra e de, por esta via (Análise
Espectral), se terem descoberto vários elementos químicos, tais como o césio, o rubídio, o tálio e o
índio, cujos nomes provêm das cores das respectivas emissões no visível (césio, de azul do céu;
rubídio, de rubi, vermelho; tálio, de talo, rebento verde; índio de índigo, corante anil). Também o
elemento hélio foi descoberto no Sol (Helios) em 1868, graças a uma risca amarela desconhecida
presente no espectro da cromosfera solar (só mais tarde se encontrou e isolou hélio na Terra).
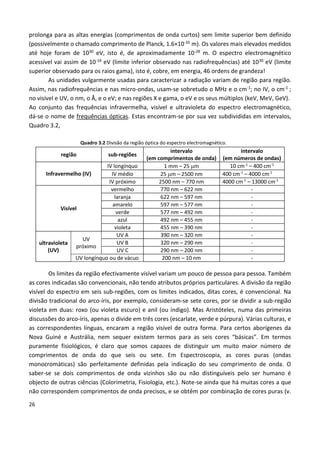
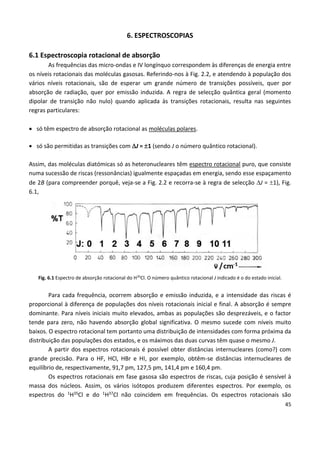
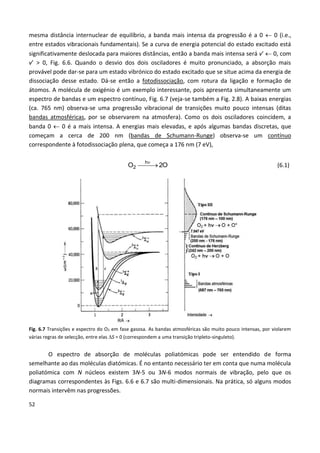
Os primeiros espectroscópios eram essencialmente constituídos por um prisma e por duas
lentes, Fig. 1.5. Foi apenas em meados do séc. XX que se substituíram os prismas por redes de
difracção, e se principiou a recorrer a outros métodos interferométricos.
Fig. 1.5 Esquema de um espectroscópio. A amostra,
colocada num fio de platina, e, é introduzida na chama, G,
de um bico de Bunsen, h. A radiação emitida passa por uma
luneta B e é refractada pelo prisma P, sendo observada
através da luneta A. A luneta C, e respectiva fonte contínua
F (vela), servem para iluminar uma escala que se sobrepõe
ao espectro. In F. F. Benevides, Noções de Physica
Moderna, 7ª ed., Lisboa, 1909. Fonseca Benevides (1835-
1911) foi professor do Instituto Industrial de Lisboa,
estabelecimento de ensino fundado em 1852 e que, em
1911, deu origem ao Instituto Superior Técnico.](https://image.slidesharecdn.com/introducaoaespectroscopia-160726061936/85/Introducao-a-espectroscopia-6-320.jpg)