Baixar para ler offline






![Introdução
● Aplicado à 2 genomas conhecidos e 1
desconhecido
● Identificando a maioria dos genes com nenhum
esforço no fechamento de gaps e resolução de
repeats
SILVA[2012]](https://image.slidesharecdn.com/seminario-chitsaz2011v4-141028203714-conversion-gate01/85/Seminario-Efficient-de-novo-assembly-of-single-cell-bacterial-genomes-from-short-read-data-sets-7-320.jpg)






![CHITSAZ[2011, p.917]](https://image.slidesharecdn.com/seminario-chitsaz2011v4-141028203714-conversion-gate01/85/Seminario-Efficient-de-novo-assembly-of-single-cell-bacterial-genomes-from-short-read-data-sets-15-320.jpg)


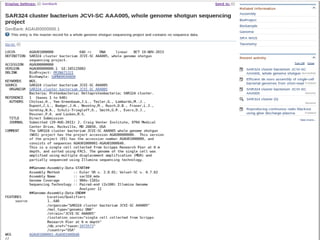
![Assembly statistic
CHITSAZ[2011, p.919]](https://image.slidesharecdn.com/seminario-chitsaz2011v4-141028203714-conversion-gate01/85/Seminario-Efficient-de-novo-assembly-of-single-cell-bacterial-genomes-from-short-read-data-sets-18-320.jpg)























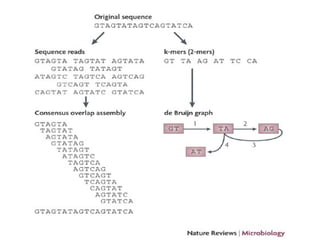
![CHITSAZ[2011, p.919]](https://image.slidesharecdn.com/seminario-chitsaz2011v4-141028203714-conversion-gate01/85/Seminario-Efficient-de-novo-assembly-of-single-cell-bacterial-genomes-from-short-read-data-sets-46-320.jpg)

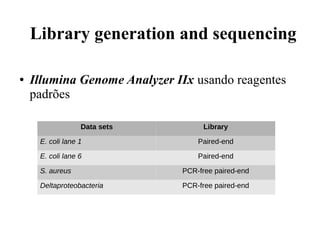
O documento descreve um método para montar genomas bacterianos a partir de dados de sequenciamento de célula única. O método combina a correção de erros do EULER-SR com a montagem do Velvet-SC para lidar com a cobertura altamente não uniforme. Isso permitiu a montagem de genomas de referência de E. coli e S. aureus, bem como um genoma desconhecido de uma Deltaproteobacteria.






















































