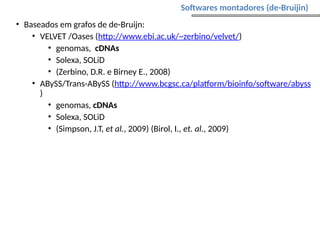
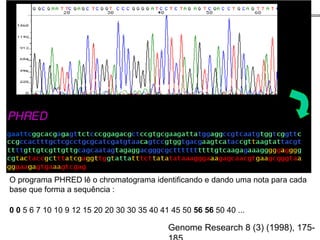
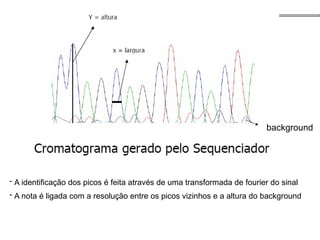
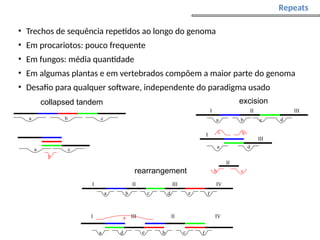
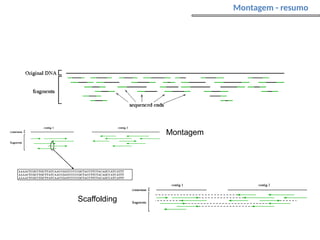
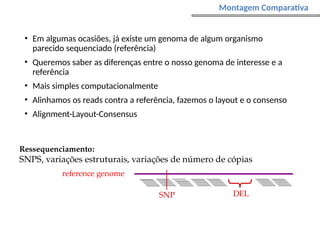
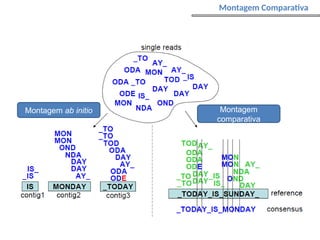
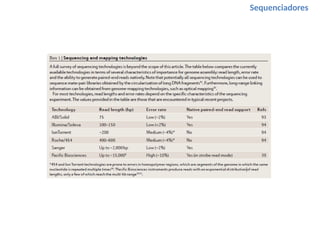
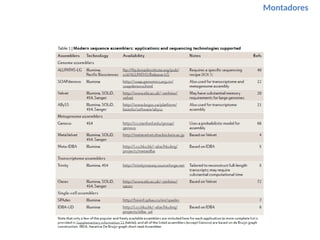
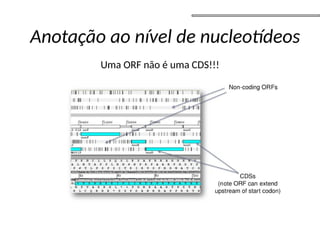
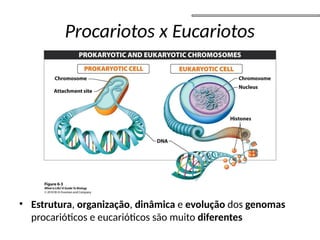
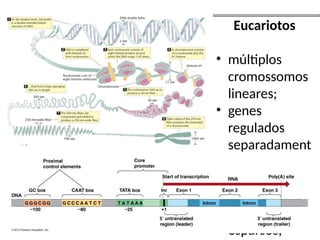
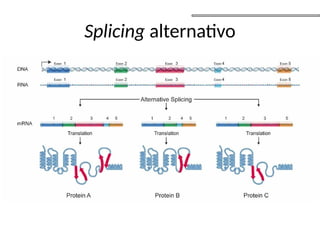
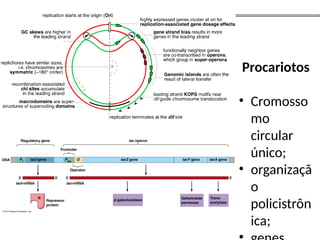
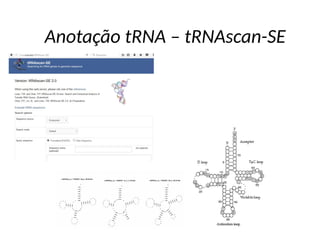
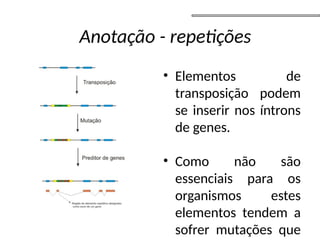
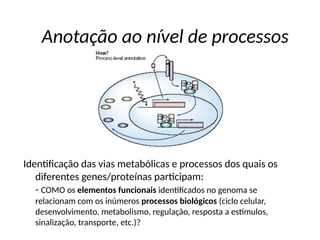
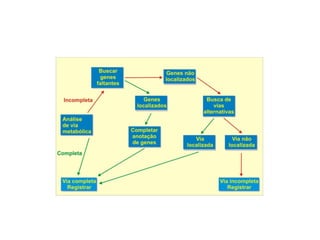
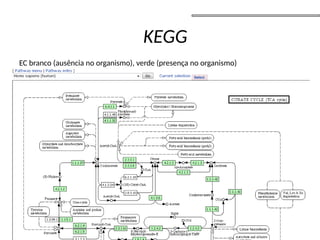
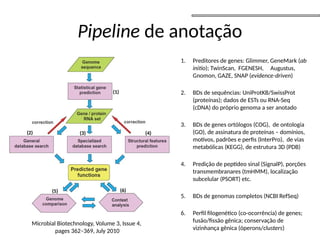
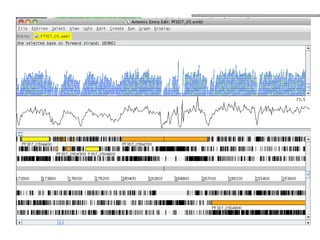
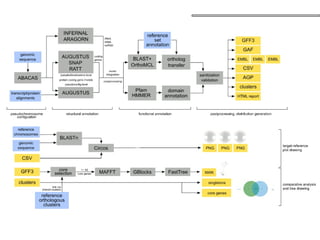
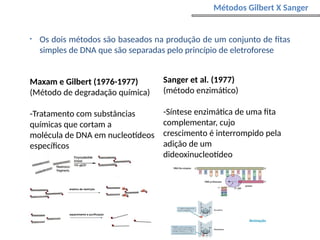
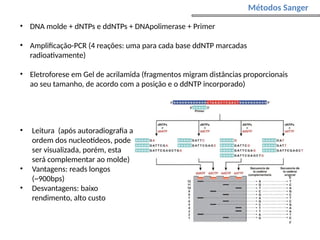
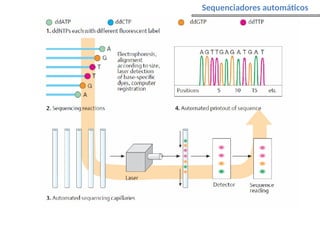
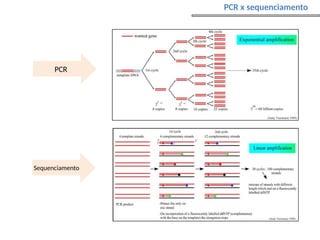
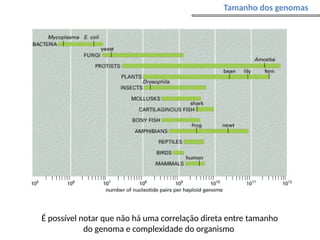
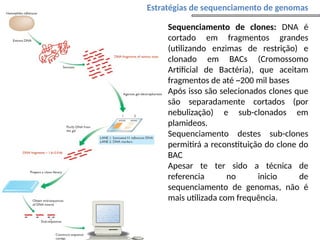
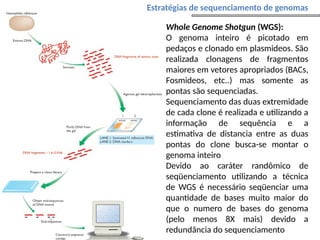
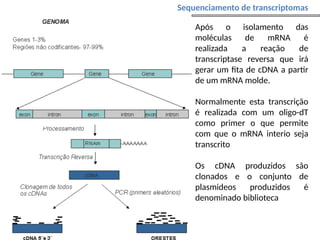
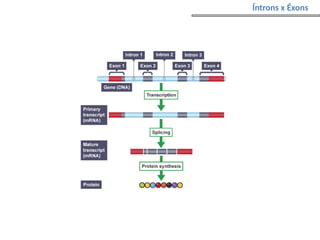
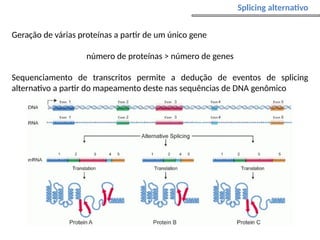
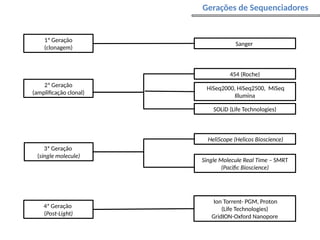
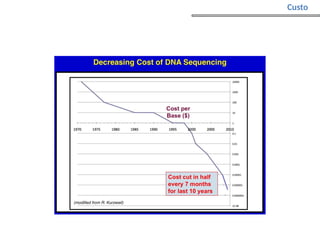
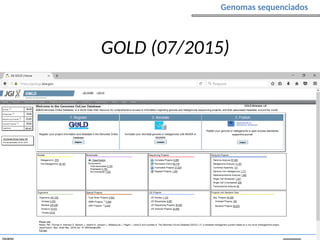
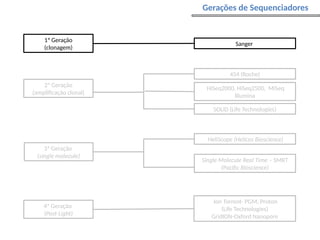
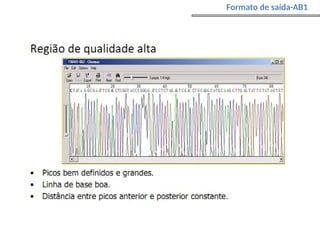
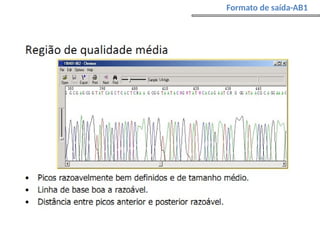
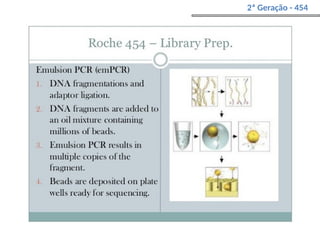
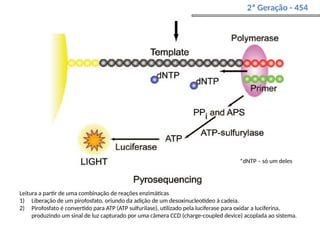
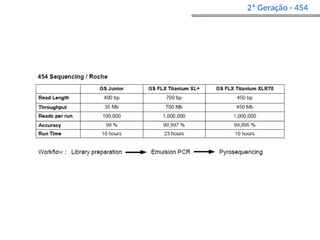
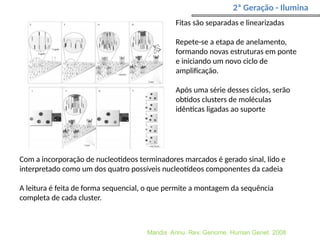
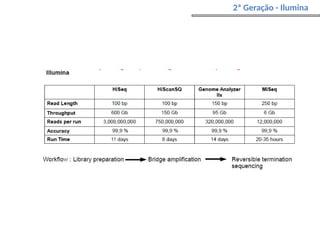
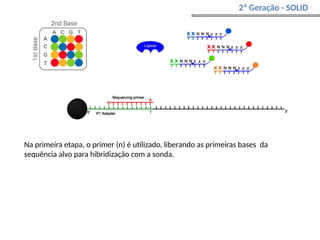
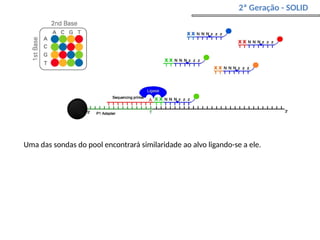
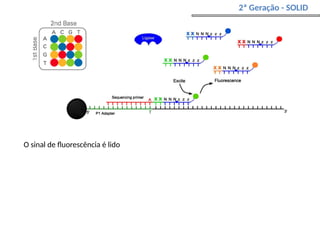
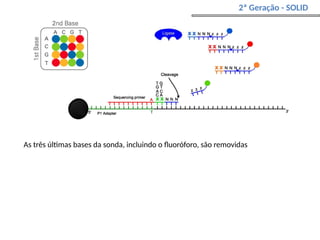
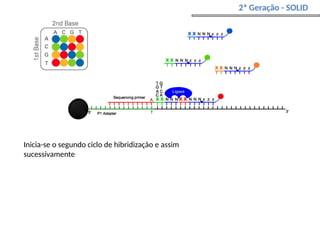
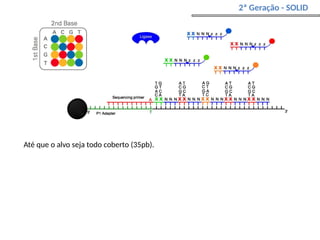
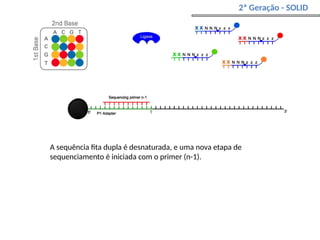
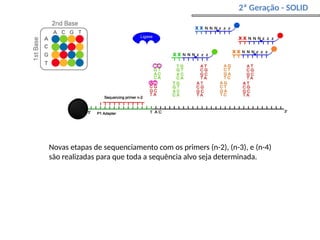
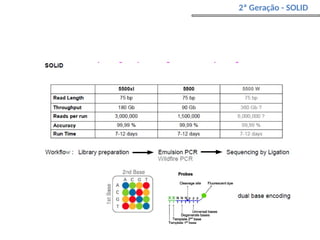
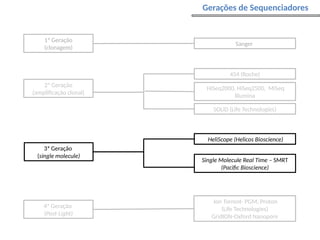
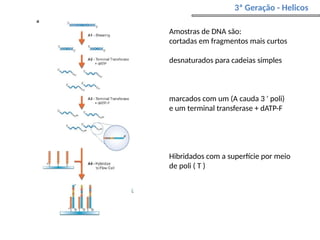
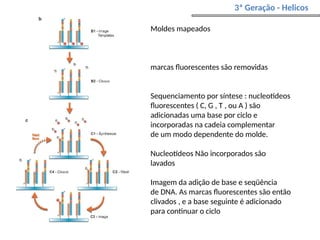
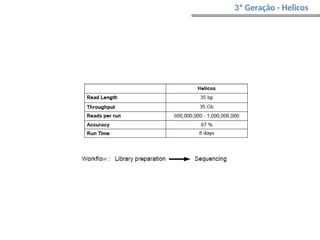
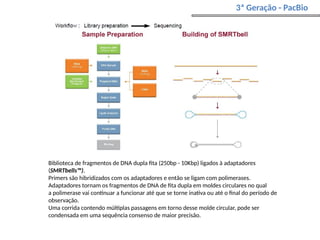
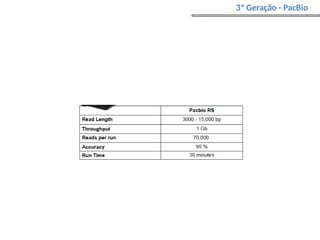
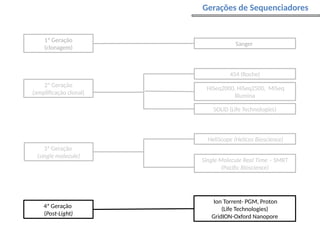
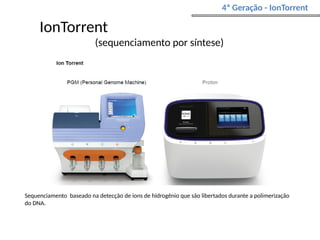
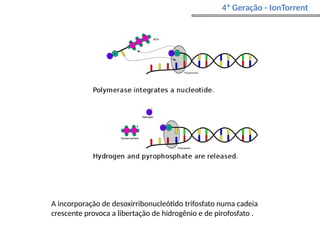
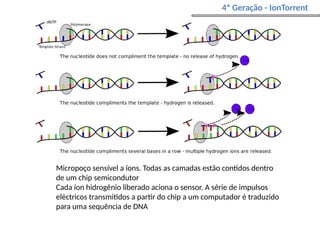
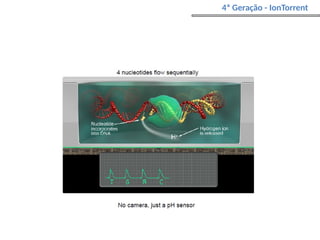
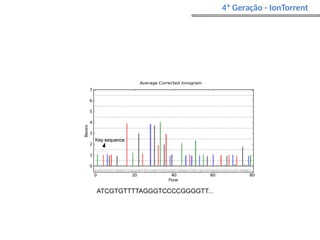
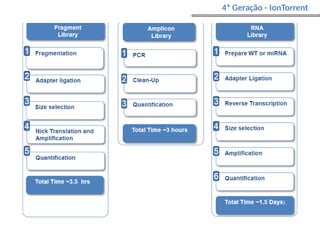
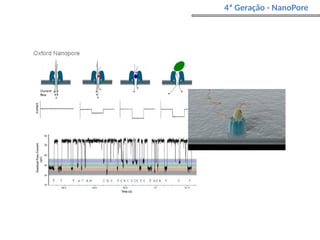
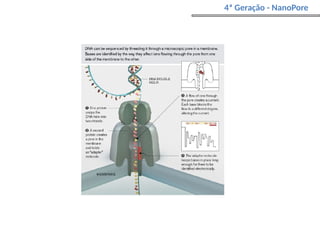
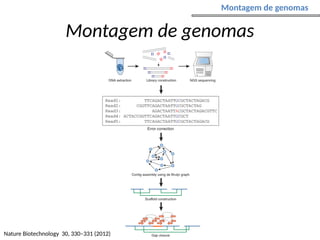
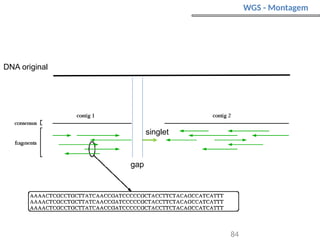
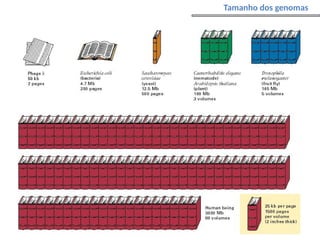
O documento aborda o sequenciamento, montagem e anotação de genomas, explicando métodos como Sanger e Next-Generation Sequencing (NGS) que revolucionaram a pesquisa genética. Discute as vantagens e desvantagens desses métodos, incluindo a geração de sequenciadores e a importância do sequenciamento de genomas para entender a composição molecular de organismos e suas evoluções. Além disso, cobre técnicas relacionadas ao sequenciamento de transcriptomas e a análise de diferentes moléculas de RNA.







































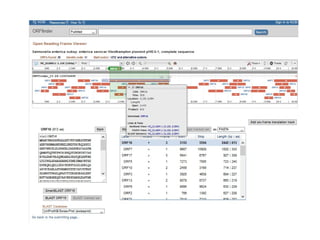
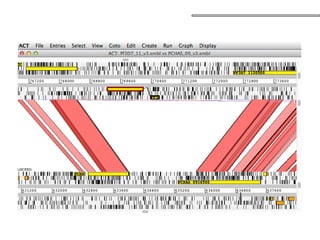
![• Três passos:
• 1º detecção de sobreposição;
• Alinhamento pareado entre todas as leituras – identificação dos pares com melhor
match (alinhamento global + heurísticas [e.g. seed & extend]);
• 2º layout dos fragmentos (montagem do contig);
• Construção e manipulação do grafo de sobreposição (Analisar/Simplificar/Limpar);
• Caminho Hamiltoniano;
• 3º decisão da sequência (montagem do consenso);
• Alinhamento Múltiplo de Sequências – normalmente baseado na pontuação dos pares
com sobreposição (sum-of-pairs ou SP);
• Realiza ajustes no layout se necessário;
• Normalmente a frequência de um nucleotídeo em determinada posição determina a
base consenso;
Caminho Hamiltoniano – caminho que
permite passar uma única vez por todos
os nós do grafo (contig) – caminho
elementar;
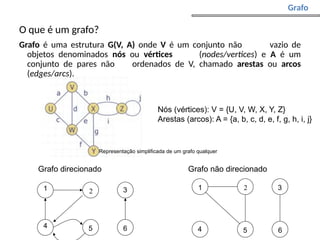
Grafo de sobreposição:
nós - reads;
arestas - sobreposições;
sobreposições não consideradas – ?caminhos alternativos?
Overlap-Layout-Consensus (OLC)](https://image.slidesharecdn.com/sequenciamentomontagemanvisa2016-241116204407-c3eba872/85/SequenciamentoMontagem_Primordios_Historico-pptx-94-320.jpg)

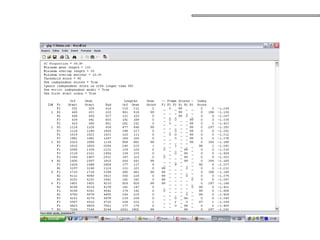
![• Grafos k-mer
• nós – todas as subsequências de tamanho k;
• arestas – todas as sobreposições (k-1 bases) entre essas subsequências
que são consecutivas na sequência original;
• Pode representar as múltiplas sequências das leituras e implicitamente
as sopreposições;
aaccgg (k-mer 4):
aacc
accg
ccgg
ccggtt (k-mer 4):
ccgg
cggt
ggtt
[Miller, et al. 2009]
Caminho Euleriano – caminho que
atravessa cada aresta uma única vez
(contig) – caminho simples;
Grafo de de-Bruijn:
nó – subsequência (k-mer);
arestas – sobreposições;
Grafos de Bruijin](https://image.slidesharecdn.com/sequenciamentomontagemanvisa2016-241116204407-c3eba872/85/SequenciamentoMontagem_Primordios_Historico-pptx-96-320.jpg)