Baixado 24 vezes






![2. Expressão Recursiva
i *δ se j=0
j *δ se i=0
OPT(i,j)
MAX [αXiYj + OPT(i -1, j -1), δ
+ OPT(i -1, j), δ + OPT(i, j -1)]
δ – gap (custo pelo espaço)
αXiYj – custo de emparelhar Xi e Yj](https://image.slidesharecdn.com/alingsequencia-pdfinal-140730195244-phpapp02/85/Alinhamento-de-Sequencia-DNA-9-320.jpg)


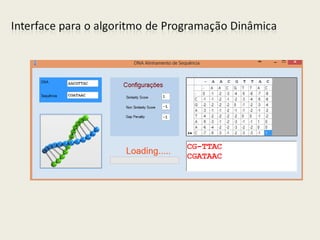
![3.2 Algoritmo Utilizando Programação Dinâmica
1. Alinhamento_PD(X,Y,δ)
2. Initialize A[i,0] = i*δ for i = 0, ..., m
3. Initialize A[0,j] = j*δ for j = 1, ..., n
4. For i = 1, ..., m
5. For j = 1, ..., n
6. A[i,j] = max (αXiYj + A[i -1, j -1], δ + A[i -1, j], δ + A[i, j -1])
7. Endfor
8. Endfor
9. Return A[m,n]](https://image.slidesharecdn.com/alingsequencia-pdfinal-140730195244-phpapp02/85/Alinhamento-de-Sequencia-DNA-12-320.jpg)


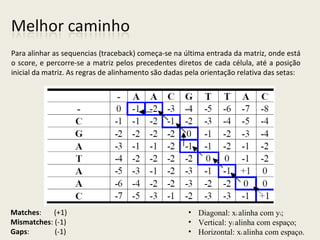
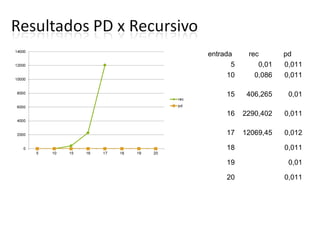



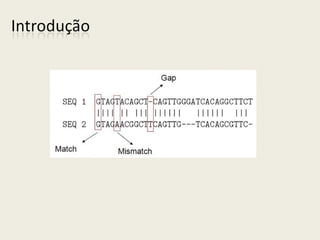
O documento discute alinhamento de sequências e como comparar duas sequências de DNA ou proteínas. Explica que alinhar sequências permite estudar evolução molecular, detectar doenças e mais. Descreve como calcular o score de um alinhamento e apresenta algoritmos recursivo e de programação dinâmica para encontrar o alinhamento ótimo.