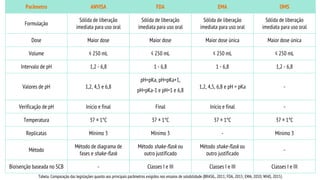
O documento discute os requisitos regulatórios para o registro de medicamentos genéricos no Brasil, incluindo estudos de equivalência farmacêutica, bioequivalência e boas práticas de fabricação. A legislação brasileira revolucionou o mercado farmacêutico ao exigir tais estudos e conceitos para comprovar a equivalência terapêutica entre medicamentos genéricos e de referência.