Baixado 86 vezes

















![RESUMO
A fitoterapia constitui uma forma de terapia medicinal que tem crescido notadamente
nestes últimos anos, criando a necessidade de garantir a eficácia, segurança e
qualidade dos medicamentos fitoterápicos. O presente estudo desenvolveu
metodologias para determinação de furanocumarinas em medicamentos fitoterápicos
empregando CLAE-UV. Foram analisados medicamentos fitoterápicos na forma de
soluções orais, cápsulas e comprimidos, os quais foram formulados, empregando
espécies de Dorstenia ou Brosimum entre outras associações. Sete deles são
indicados para tratamento de distúrbios menstruais ou menopausa. Três deles são
utilizados para tratamento do vitiligo. Apenas um dos medicamentos possui
informação sobre a presença de uma furanocumarina em sua composição nas bulas
ou folhetos. Os métodos otimizados foram validados segundo as normas da
International Conference on Harmonization, mostrando boa especificidade, acurácia,
precisão e linearidade. O estudo interequipamento utilizando CG-DIC para soluções
orais e CG-EM para cápsulas e comprimidos, não revelou diferenças estatísticas
significativas. Devido a grande variedade das formulações dos medicamentos
utilizados no desenvolvimento e validação dos métodos, sugere-se que estes
possam ser utilizados para análise de psoraleno, bergapteno e 5-[3-(4,5-Diidro-5,5-
dimetil-4-oxo-2-furanil)-butoxi]-7H-furo[3-2-g][1] benzopiran-7-ona em outras
formulações de fitoterápicos presentes no mercado. O estudo do perfil
cromatográfico dos medicamentos foi realizado em condições isocráticas e em
gradiente por CLAE-UV. Foram determinadas furanocumarinas em nove
medicamentos analisados, o que oferece riscos à saúde pública devido ao
desconhecimento da presença destas substâncias, na maioria destes produtos.
Além disso, a furanocumarina encontrada em maior quantidade nos medicamentos
estudados foi o psoraleno, considerado mais tóxico que a xantotoxina, normalmente
prescrita pelos médicos para tratamento de doenças de pele. O uso de fitoterápicos
contendo furanocumarinas no tratamento de distúrbios menstruais pode ser
considerado de alto risco se for considerada a relação risco-benefício, pois se
acredita que sejam indicados pelas propriedades anticoagulantes das cumarinas
presentes nas plantas da formulação. Realizou-se também o estudo do perfil
cromatográfico e dosagem de furanocumarinas em plantas presentes nas
formulações dos medicamentos, utilizando tanto amostras adquiridas no comércio
como amostras identificadas.
xii](https://image.slidesharecdn.com/tesecompletacomtudo130404-090912175819-phpapp02/85/Minha-dissertacao-2004-17-320.jpg)
![ABSTRACT
The phytoterapy constitutes a form of medical therapy that notably have
been growing in the last years, creating the necessity to guarantee the effectiveness,
security and quality of phytomedicines. The present study has the development of
methodologies for the determination HPLC of furanocoumarins in phytomedicines.
Four oral solutions, three capsules and three tablets, all formulated using species of
Dorstenia or Brosimum among others associations, were analyzed. Seven of them
are indicated for the treatment of menstrual irregularities and disturbances related to
menopause. The other three are used for the treatment of vitiligo. Only one of the
medicines showed the information on the presence of furanocoumarin in its
composition. The optimized methods were validated according to the International
Conference of Harmonization, showing good specificity, accuracy, the inter- and
intra-day precision and linearity. The inter-equipment study was performed using GC-
FID for oral solutions and GC-MS for capsules and tablets. They did not displayed
significant statistic differences. Due to the great variety in the formulations of the
medicines used in the development and validation of the methods, it is suggested
that they can be used for analysis of psoralen, bergapten and 5-[3-(4,5-Dihydro-5,5-
dimethyl-4-oxo-2-furanyl)-butoxy]-7H-furo[3-2-g][1] benzopyran-7-one in the
formulation of other phytomedicines found at commerce. The study of the
chromatographic profile of medicines was evaluated in isocratic and gradient runs by
HPLC-UV. Furanocoumarins were detected in nine of the medicines analyzed. The
lack of knowledge of the presence of these substances in most of the analyzed
products offers risks to the public health. Moreover, psoralen was the furanocoumarin
found in the largest amount. Psoralen is considered more toxic than xanthotoxin,
which is normally prescribed by doctors on the treatment of skin illnesses. If we
consider the relation risk-benefit on the treatment of menstrual riots, the use of
phytomedicines containing furanocoumarins can be of high risk, since it is believed
that they are indicated because of the anticoagulation properties of coumarins found
on the plant’s formulation. The study of the chromatographic profile and dosage of
furanocoumarins in the plants found at the medicines’ formulation was also made,
using identified samples and samples acquired at commerce.
xiii](https://image.slidesharecdn.com/tesecompletacomtudo130404-090912175819-phpapp02/85/Minha-dissertacao-2004-18-320.jpg)







![Introdução
das espécies africanas D.psilurus45 e D.barnimiana46 e D. poinsettifolia47.
Furanocumarinas também foram encontradas em galhos de D. prorepens35 e D.
gigas34 e em folhas de D. gigas34.
Estruturalmente, as furanocumarinas em Dorstenia apresentam-se pouco
diversificadas, havendo predomínio de furanocumarinas angulares ou lineares
simples tais como bergapteno, psoraleno, isopimpinelina, pimpinelina, isobergapteno
etc. As exceções são três furanocumarinas preniladas encontradas em D. cayapiaa -
5-[3-(4,5-diidro-5,5-dimetil-4-oxo-2-furanil)butoxi-7H-furo[3-2-g][1]benzopiran-7-ona,
D.brasiliensis – 5-[3-(4,5-diidro-5,5-dimetil-4-oxo-2-furanil)butoxi-7H-furo[3-2-
g][1]benzopiran-7-ona (Figura 3, estrutura a) e o análogo insaturado 5-[[3-(4,5-diidro-
5,5-dimetil-4-oxo-2-furanil) 2-butenil-]oxi]-7H-furo[3-2-g][1]benzopiran-7-ona (Figura
3, estrutura b)41 e D.contrajerva – 4-[[3-(4,5-diidro-5,5-dimetil-4-oxo-2-furanil)-
butil]oxi]-7H-furo[3,2-g][1]benzopiran-7-ona43.
Figura 3 – Furanocumarinas preniladas encontradas em Dorstenia brasiliensis.
Estudos fitoquímicos do gênero relatam ainda a ocorrência de
cardenolídios48, triterpenos37, ácidos graxos42, esteróides41, diferentes
benzofuranos46 e vários flavonóides geranilados e prenilados49,50.
Desenvolvimento e Validação de Metodologias para Determinação de Furanocumarinas em 8
Medicamentos Fitoterápicos.](https://image.slidesharecdn.com/tesecompletacomtudo130404-090912175819-phpapp02/85/Minha-dissertacao-2004-26-320.jpg)

![Introdução
amenorréia55,57 e na solidificação de ossos fraturados52, em mistura com a taiuiá
(Trianosperma tayuya)55.
Há apenas dois trabalhos sobre o isolamento de furanocumarinas da
espécie41,56 que identificaram psoraleno, bergapteno, 5-[3-(4,5-Diidro-5,5-dimetil-4-
oxo-2-furanil)-butoxi]-7H-furo[3-2-g][1] benzopiran-7-ona, 5-[[3-(4,5-diidro-5,5-dimetil-
4-oxo-2-furanil)-2-butenil-]oxi]-7h-furo[3-2-g][1]benzopiran-7-ona), (2’S,3’R)-3’-
hydroximarmesina 4’-O-β-D-glucopiranosida, (2’S)-marmesin 4’-O-α-L-
rhamnopiranosil (1→6)-O-β-D-glucopiranosida, (2’S,3’R)-3’-hydroximarmesina, 2’-(1”-
hidroxi-1”-metiletil)-psoraleno, 7-hydroxicumarina, 3-O-β-glucosilsitosterol, sucrose,
esteróides, diterpenóides e triterpenóides das raízes/rizomas da espécie. Os
triterpenóides (ácidos dorstênicos A e B) isolados de D. brasiliensis, apresentaram
moderada citoxicidade sobre células da leucemia (L-1210 e HL-60)56.
O nome popular “carapiá” também é encontrado referindo-se a outras
duas espécies: Cordia superba Cham.58 da família Boraginaceae, encontrada no Rio
de Janeiro, Minas Gerais e São Paulo e Sida macrodon52, da família das Malváceas.
A primeira espécie também pode ser encontrada com os nomes de árvore de ranho
e grão de galo e a segunda, como malva do campo.
1.3.2. Brosimum gaudichaudii
Brosimum gaudichaudii é uma espécie conhecida como “mamica de
cadela”. Utilizada em doenças de pele devido às furanocumarinas59. É nativa do
cerrado brasileiro, com uma ampla distribuição por todo o Brasil central 60. Análises
da casca da raiz de B. gaudichaudii identificaram derivados dos ácidos cinâmicos e
diidrocinâmico, dez cumarinas (dentre elas psoraleno, bergapteno, xantiletina,
luvangetina e (+)-(2’S,3’R)-1’-hidroximarmesina) (Figura 4), uma chalcona, os
esteróides: β-sitosterol e 3-β-O-β-D-glicopiranosil- β-sitosterol e o triterpeno β-
amirina61,62.
Desenvolvimento e Validação de Metodologias para Determinação de Furanocumarinas em 10
Medicamentos Fitoterápicos.](https://image.slidesharecdn.com/tesecompletacomtudo130404-090912175819-phpapp02/85/Minha-dissertacao-2004-28-320.jpg)





















![Parte Experimental
3. PARTE EXPERIMENTAL
3.1. Materiais e Equipamentos Utilizados
3.1.1. Materiais utilizados
Nas análises por cromatografia em camada delgada (CCD) foram
utilizadas placas de sílica gel 60 f 254 da Merck. Os padrões de psoraleno,
bergapteno, isopimpinelina, isobergapteno e 5-[3-(4,5-Diidro-5,5-dimetil-4-oxo-2-
furanil)-butoxi]-7H-furo[3-2-g][1] benzopiran-7-ona (denominado no trabalho como
DT) utilizados foram isolados e purificados por nosso grupo de pesquisa (psoraleno
98,89%, bergapteno 99,01% , DT 97,45% , pimpinelina 98,01% e isopimpinelina
97,34% de pureza) empregando técnicas cromatográficas e ressonância magnética
nuclear.
3.1.2. Solventes utilizados
Os solventes orgânicos utilizados nas extrações de plantas e como
eluentes nas análises em CCD foram solventes de grau P.A. Os solventes
empregados no desenvolvimento das metodologias para os medicamentos
fitoterápicos e aqueles empregados em CLAE-UV, CG-DIC e CG-EM foram grau
CLAE (Darmstadt, Germânia). A água utilizada foi tratada pelo sistema Milli-Q
(Millipore).
3.1.3. Equipamentos utilizados
Ultra-som de mesa marca Thornton, tipo T14 modelo C/T;
Luz UV de comprimentos de onda 254 nm e 366 nm;
Centrífuga: Excelsa Baby I modelo 206;
Balança analítica: Scientech SA120 (precisão 0,001g).
Desenvolvimento e Validação de Metodologias para Determinação de Furanocumarinas em 33
Medicamentos Fitoterápicos.](https://image.slidesharecdn.com/tesecompletacomtudo130404-090912175819-phpapp02/85/Minha-dissertacao-2004-51-320.jpg)



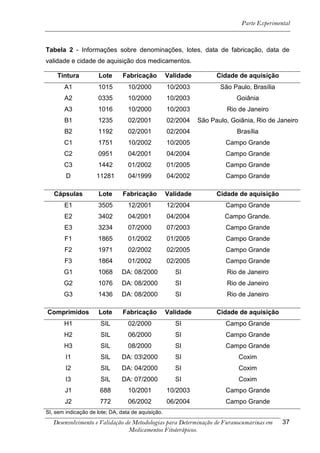












![Resultados e Discussão
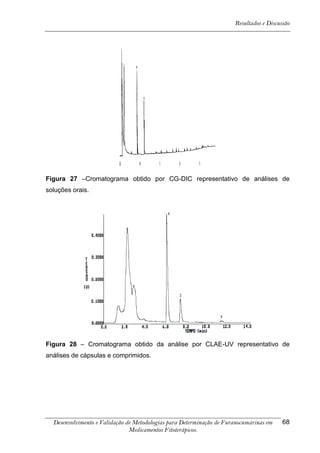
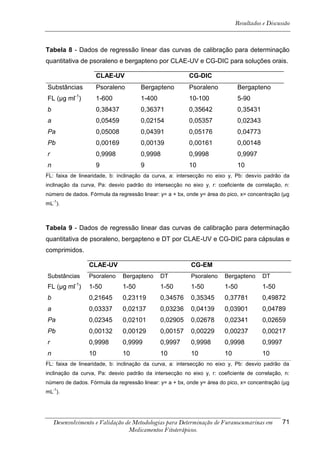
4. RESULTADOS E DISCUSSÃO
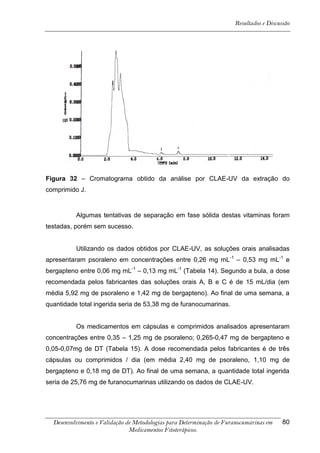
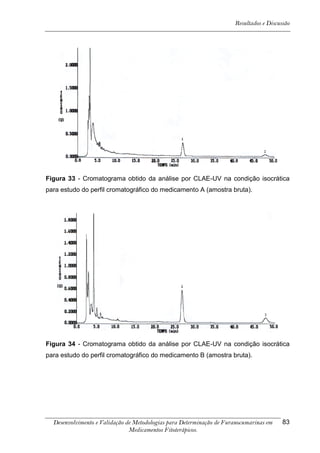
Os cromatogramas obtidos por CLAE-UV em todas as condições
cromatográficas, por CG-DIC e por CG-EM possuem os números 1, 2 e 3 indicando
psoraleno, bergapteno e DT (Figura 10), respectivamente, quando presentes nas
amostras.
*
Figura 10 - Estrutura das substâncias analisadas
A análise em CLAE-UV, na condição empregada para quantificação,
mostrou uma boa separação dos picos dos compostos de interesse na linha de base
que poderiam ser analisados num intervalo de tempo satisfatório, menor que doze
minutos para soluções orais, cápsulas e comprimidos (Tabela 4). A análise em GC-
FID e CG-EM também mostrou uma boa separação dos picos dos compostos de
interesse na linha de base que poderiam ser analisados num intervalo de tempo
satisfatório, menor que 17 minutos (Tabela 4). As amostras A, B, E, G e I, que não
continham DT, puderam ser analisadas em tempo menor que 8 minutos em todos os
equipamentos.
*
5-[3-(4,5-Diidro-5,5-dimetil-4-oxo-2-furanil)-butoxi]-7H-furo[3-2-g][1] benzopiran-7-ona
Desenvolvimento e Validação de Metodologias para Determinação de Furanocumarinas em 50
Medicamentos Fitoterápicos.](https://image.slidesharecdn.com/tesecompletacomtudo130404-090912175819-phpapp02/85/Minha-dissertacao-2004-68-320.jpg)

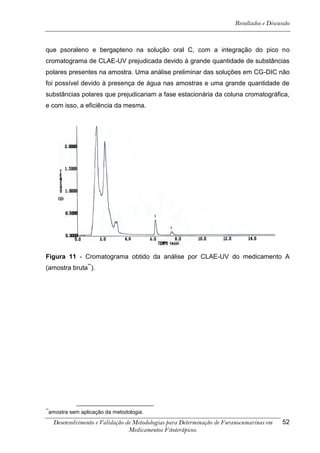
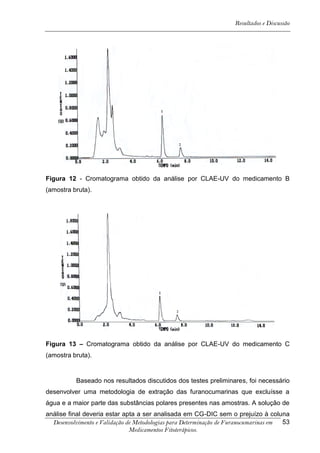

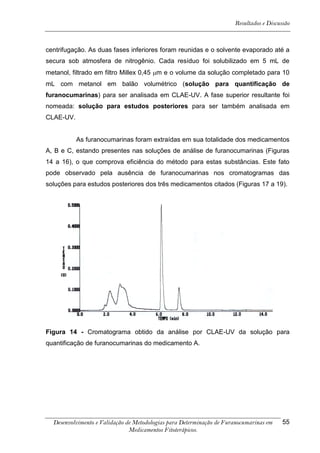
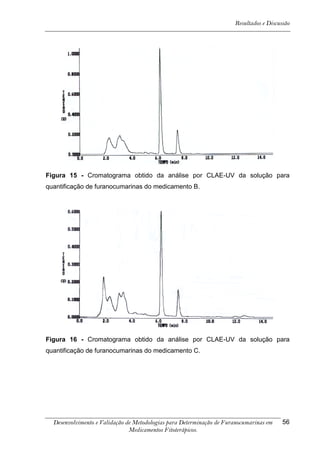
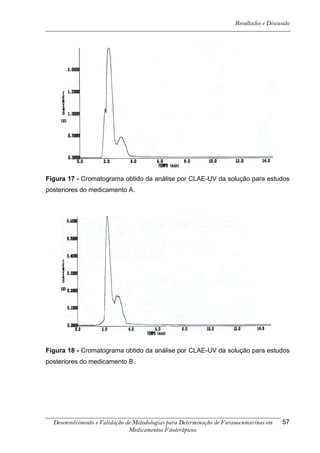
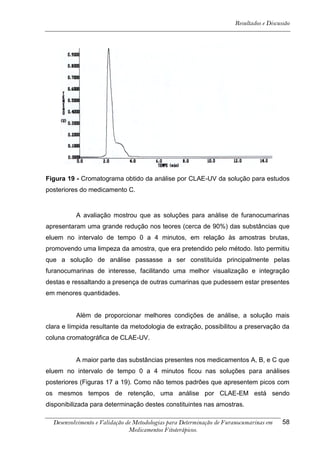
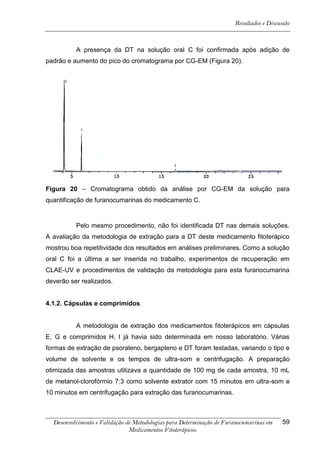

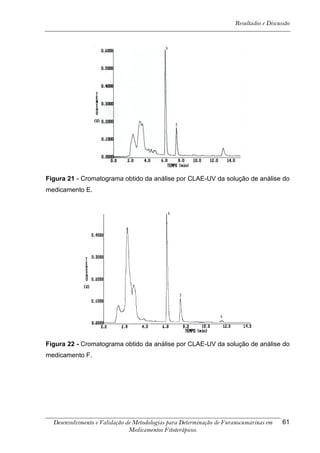
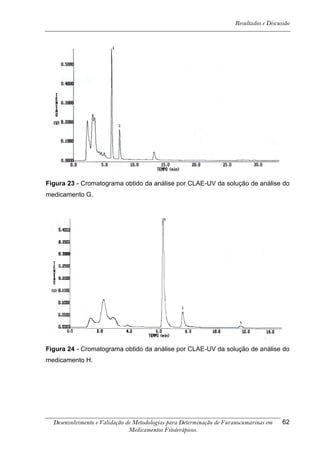


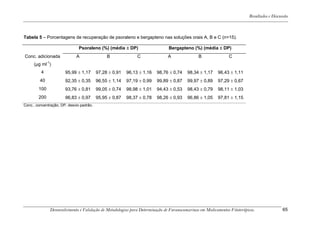





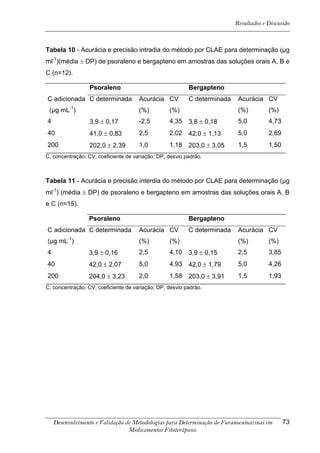
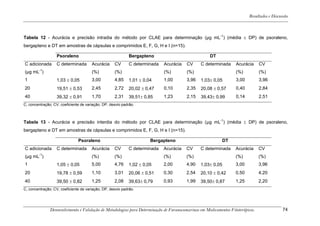


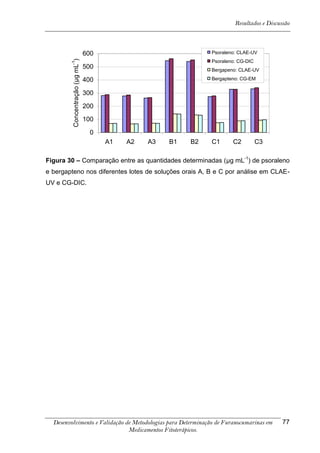
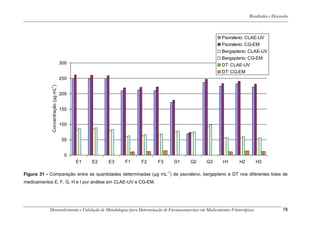



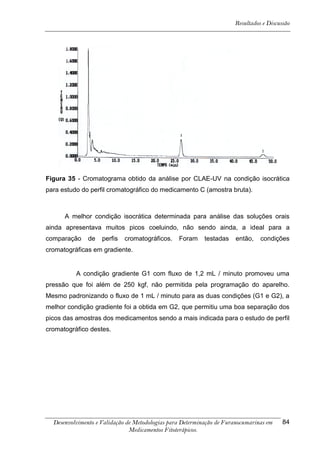
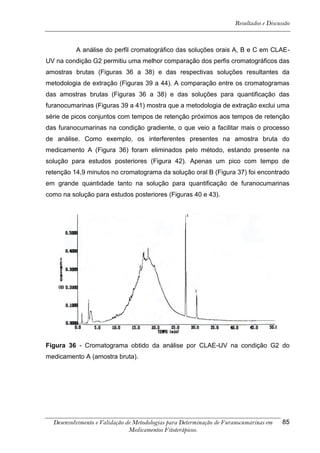
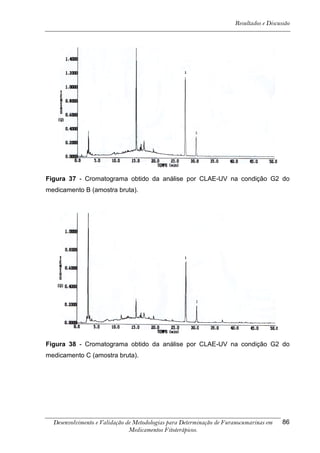
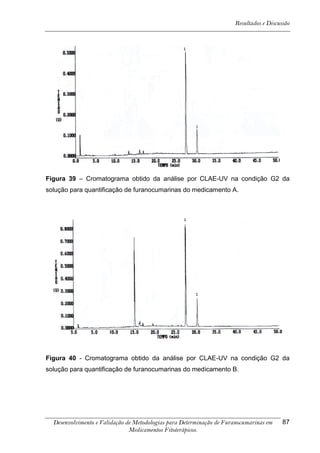
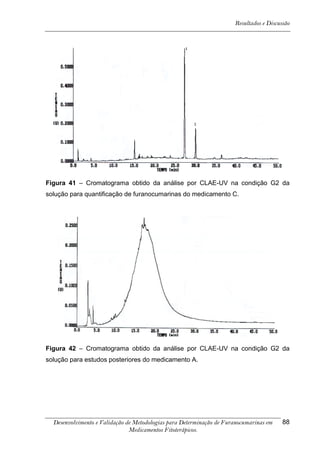


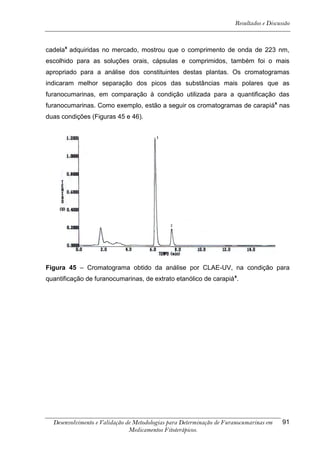

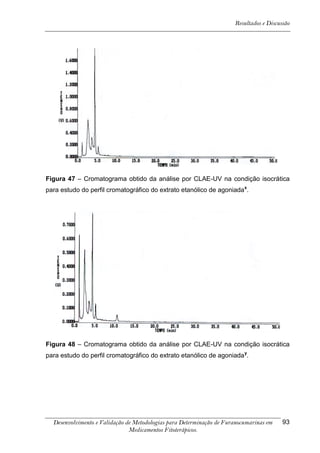
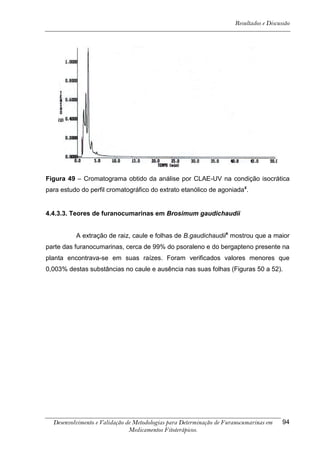
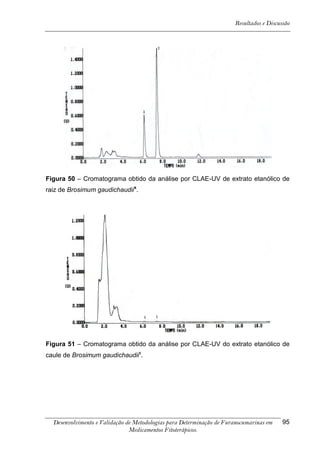


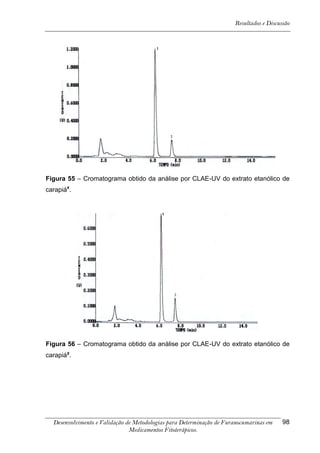
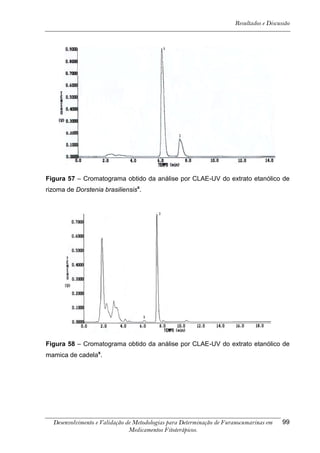
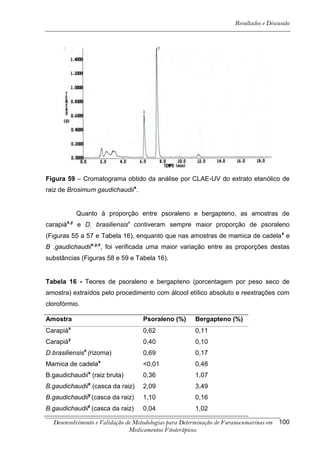


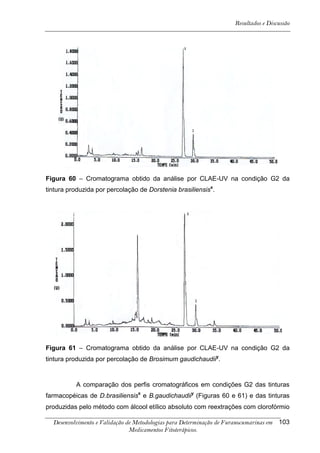
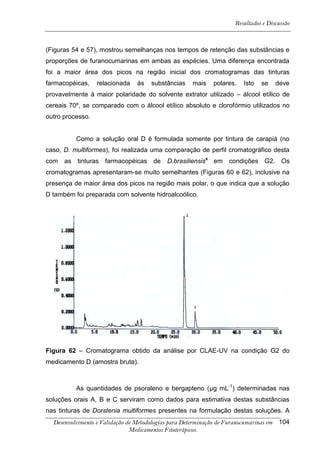

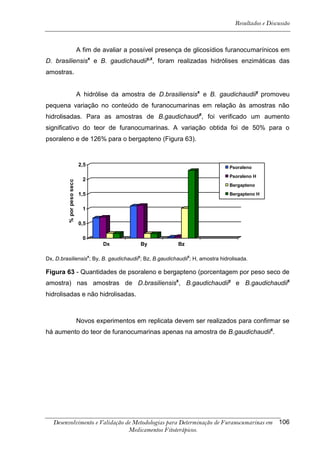














Este trabalho apresenta o desenvolvimento e validação de metodologias para determinação de furanocumarinas em medicamentos fitoterápicos por cromatografia líquida de alta eficiência com detecção por UV. Foram desenvolvidas metodologias para soluções orais, cápsulas e comprimidos, validadas por parâmetros como recuperação, especificidade, limites de detecção e quantificação, estabilidade, curva de calibração e precisão. Estudos preliminares com plantas presentes nos medicamentos foram realizados para identificação












![Processo de refino_petrobras_[1]](https://cdn.slidesharecdn.com/ss_thumbnails/processoderefinopetrobras1-131017203645-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)





![[Apostila] termodin mica](https://cdn.slidesharecdn.com/ss_thumbnails/apostilatermodinmica-100824125426-phpapp01-thumbnail.jpg?width=640&height=640&fit=bounds)



















































