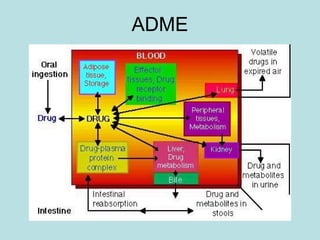
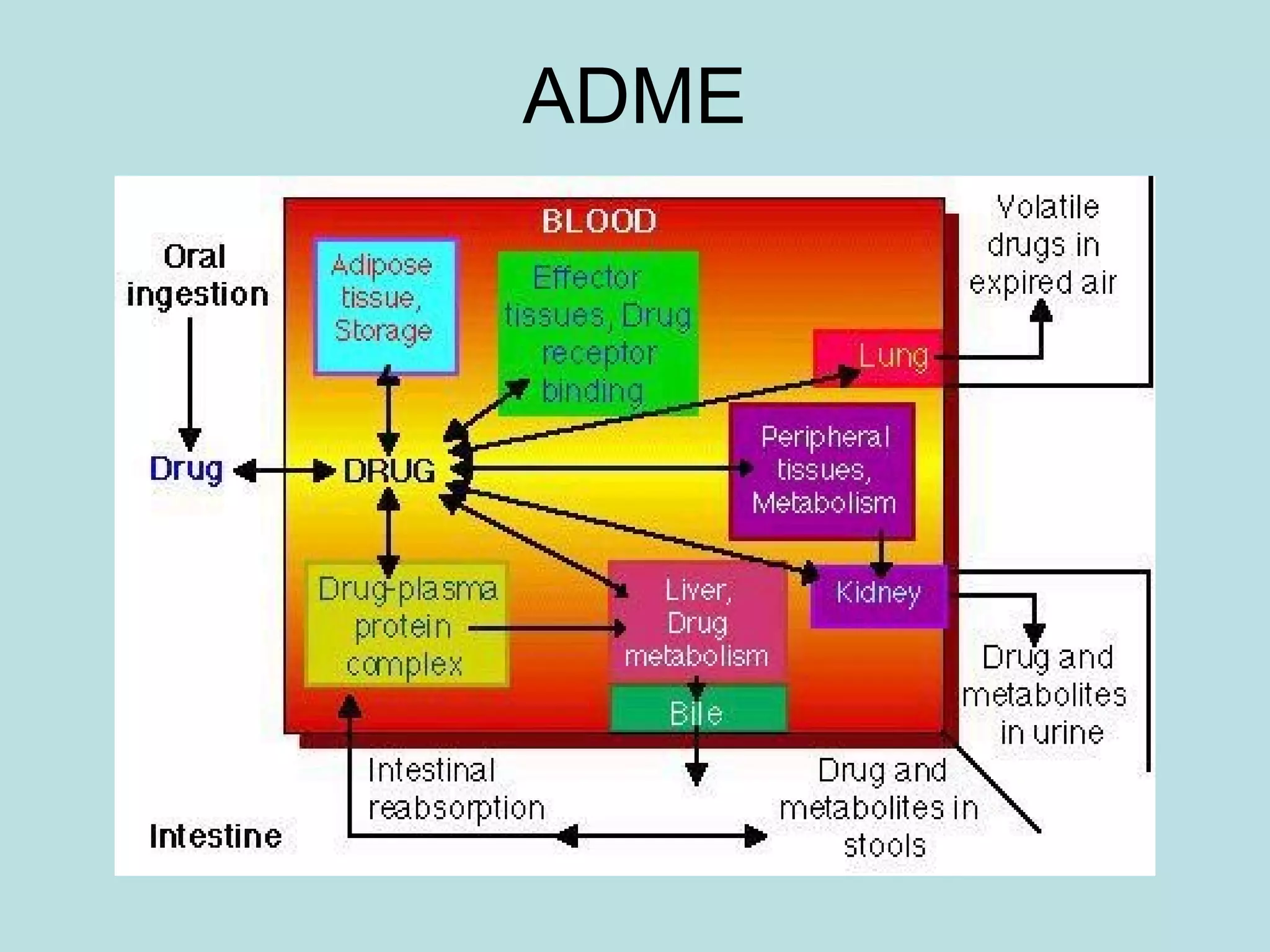
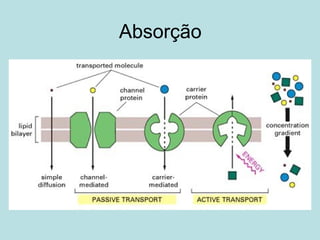
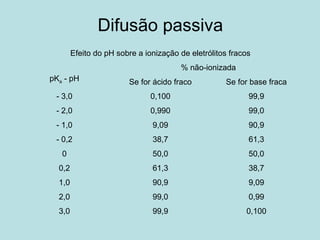
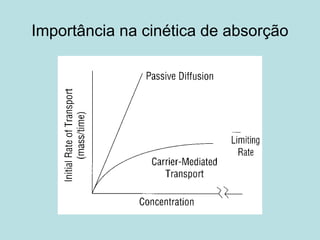
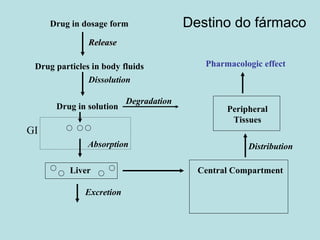
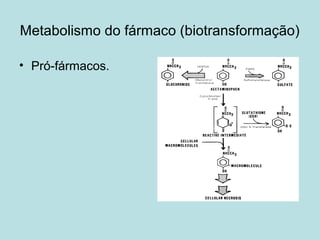
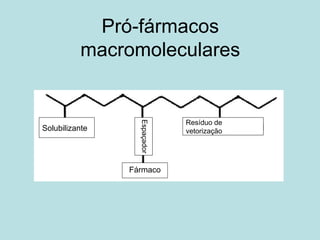
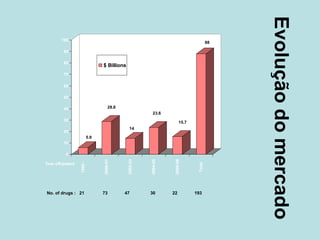
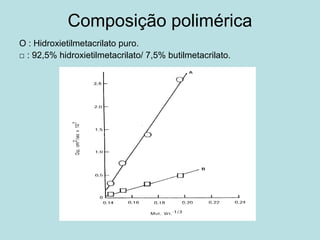
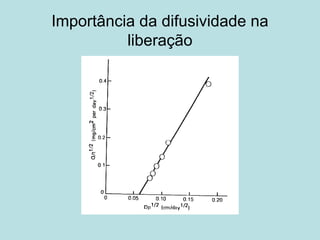
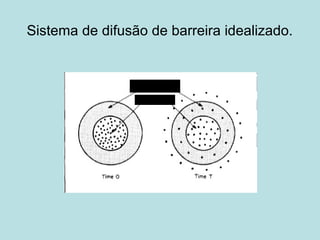
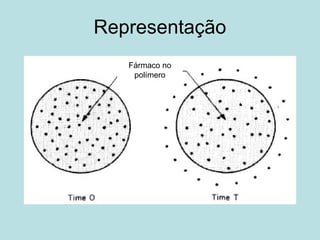
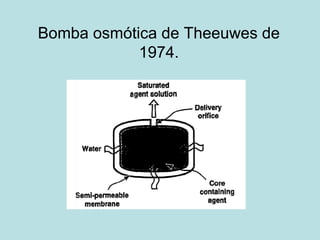
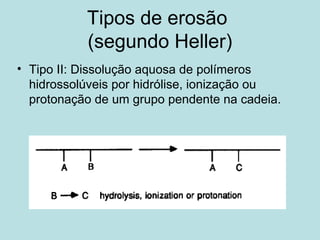
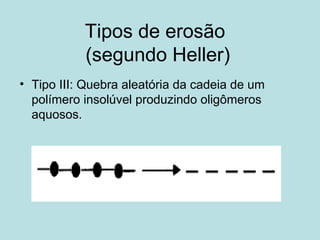
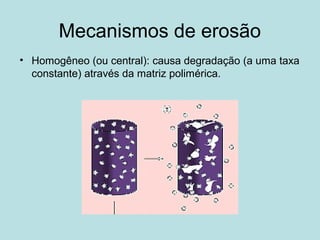
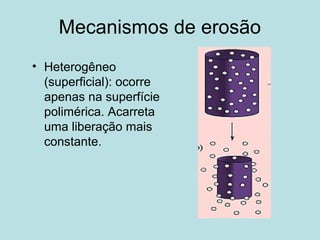
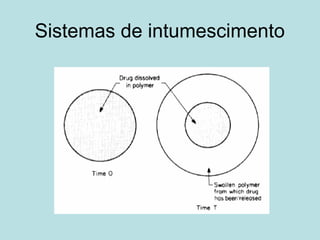
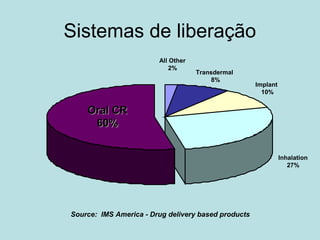
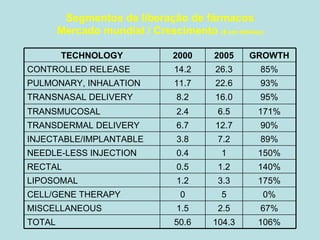
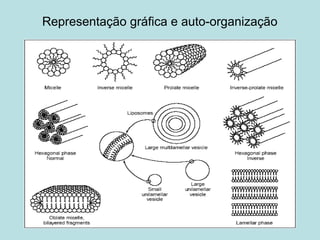
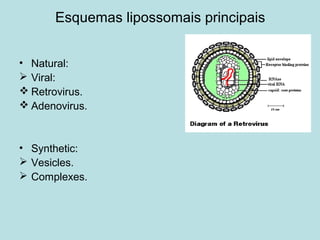
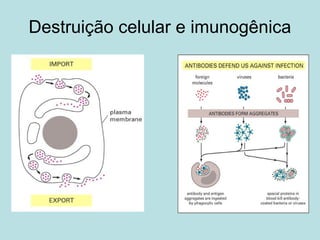
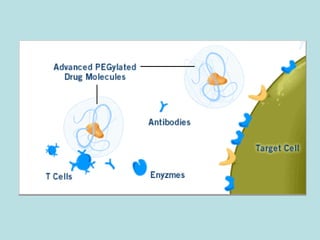
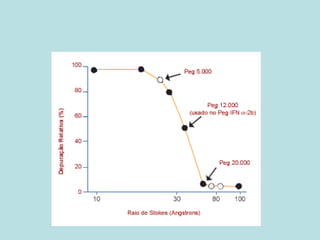
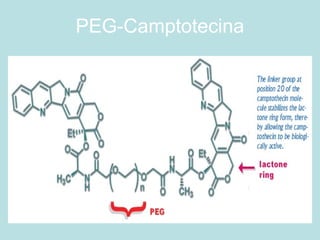
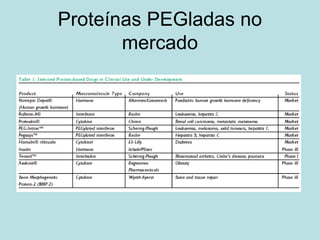
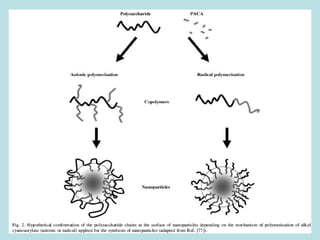
O documento descreve os principais mecanismos de absorção de fármacos, incluindo difusão passiva e transporte ativo através de membranas biológicas. Também discute diferentes sistemas de liberação controlada de fármacos, como sistemas controlados por difusão, solvente, degradação química e intumescimento. Vários fatores que afetam a cinética de absorção e liberação são detalhados, como propriedades físico-químicas dos fármacos e polímeros.