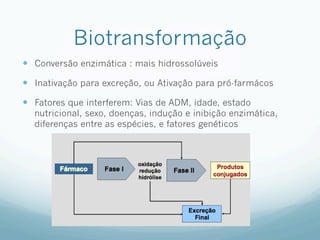
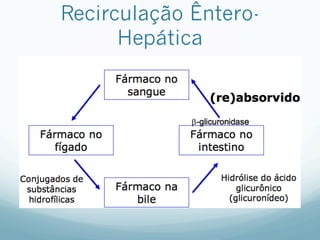
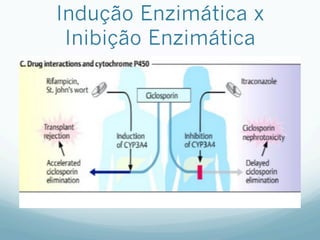
Este documento resume os principais mecanismos de transporte de fármacos através das membranas biológicas, como difusão, difusão facilitada e transporte ativo, além de conceitos sobre receptores farmacológicos, transdução de sinais, regulação e teorias sobre ação dos fármacos. Também aborda brevemente os processos de absorção, distribuição, biotransformação e excreção de fármacos no organismo.


![Os Mecanismos de
Transporte
Difusão
Sem gasto energético
A favor do gradiente
de [ ]
Interrompe-se quando
ambos lados estão em
equilíbrio
Difusão
Facilitada
Moléculas
carreadoras
Uni, Anti e
Simportadoras
Canais
Muito mais rápido
Depende da mudança
de potencial elétrico
na membrana e da
substância
indutora(ligante)
Transporte
Ativo
Gasto enérgetico
( energia é a mudança
conformacional)
Contra o gradiente de
[ ]
Saturação dos
transportadores](https://image.slidesharecdn.com/monitoria-p1-220718000610-b501a643/85/Monitoria-P1-pdf-3-320.jpg)
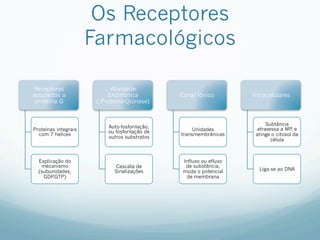
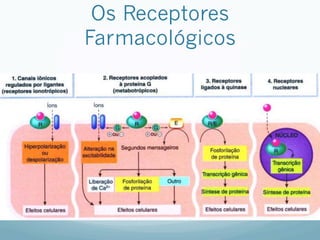



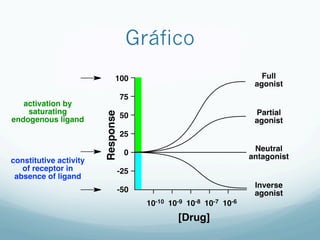
![Especificidade
— É a capacidade de produzir um ÚNICO efeito.
— Ligada á [ ] à porém tomar cuidado: pode atingir
efeitos secundários em maiores concentrações à
pode perder a especificidade
— Nenhum fármaco é 100 % específico à efeitos
colaterais](https://image.slidesharecdn.com/monitoria-p1-220718000610-b501a643/85/Monitoria-P1-pdf-11-320.jpg)
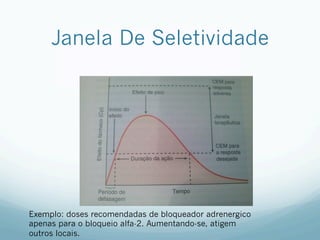
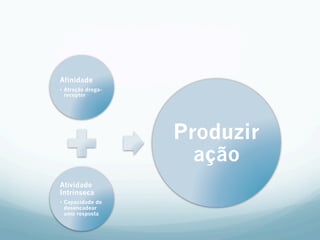
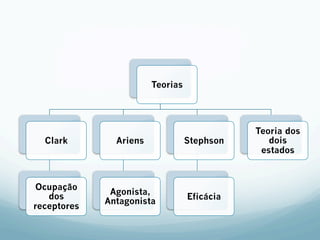



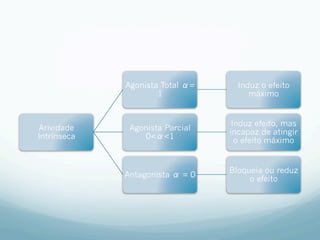

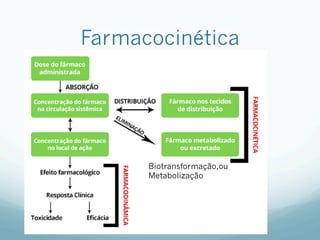
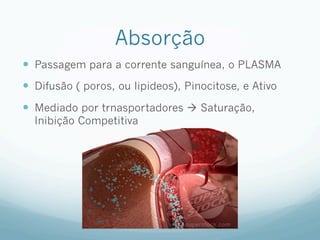
![Absorção
— Grau de ionização
— Fármacos são melhores absorvidos na sua FORMA
MOLECULAR
— As formas ionizadas possuem lipossolubilidade baixa
— Fatores que influenciam
— Área, Gradiente de [ ], Lipossolubilidade, Polaridade (apolar
não tem interação),Ionização (forma molecular é melhor
absorvido) e PM (menor tamanho e volume é melhor
absorvido)
— Relacionado ao indíviduo: tempo de trânsito
gastrointestinal, presença de alimentos, e fluxo sanguíneo
— Relacionado as vias de administração: IV não tem efeito de
primeira passagem, 100 % biodisponível.](https://image.slidesharecdn.com/monitoria-p1-220718000610-b501a643/85/Monitoria-P1-pdf-23-320.jpg)