Baixado 13 vezes



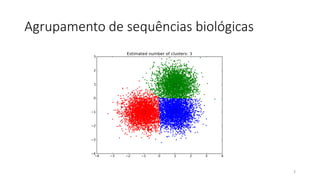



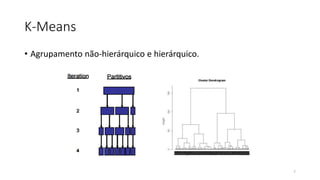






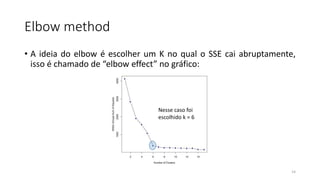







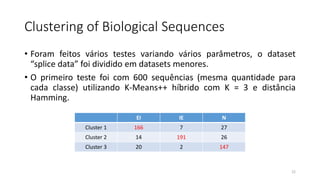
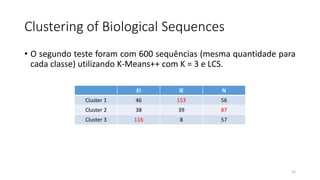
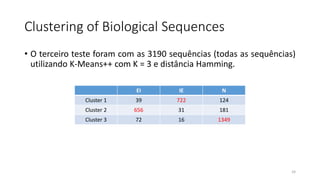
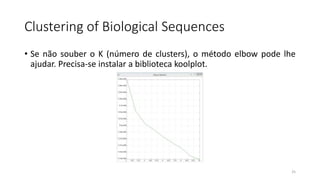





O documento discute técnicas de agrupamento de sequências biológicas, incluindo K-Means, K-Means++, método do cotovelo e um algoritmo híbrido. Ele também apresenta um projeto open-source para agrupar sequências usando essas técnicas e mede a acurácia em diferentes conjuntos de dados.













































