Público
Leis Federais elegislação farmacêutica
Yslla Milla
Farmacêutica CRF/MA 8337
“primum, non nocere” (“antes de tudo, não causarei dano ao
paciente”)
Público
Conselhos e outrasautarquias relacionadas à
farmácia
Conselho Regional de Farmácia CRF
Conselho Federal de Farmácia CFF
Agência Nacional de Vigilância Sanitária (ANVISA)
Vigilância Sanitária (VISA)
Organização Mundial da Saúde (OMS)
Agência Reguladora de Medicamentos FDA
4.
Público
CRF &CFF
Os conselhos federais e regionais foram criados com o objetivo de
regulamentar, padronizar e fiscalizar o exercício prossional das atividades
farmacêuticas das instituições e dos prossionais inscritos nestes conselhos.
Estes órgãos existem com o intuito de garantir a qualidade dos serviços
farmacêuticos prestados à saúde e os interesses coletivos e individuais dos
prossionais.
5.
ANVISA
BR: agênciaresponsável pela regulação de medicamentos;
Criada pela Lei nº 9.782, de 26 de janeiro de 1999;
É uma autarquia com autonomia financeira e ligada ao Ministério da
Saúde(MS);
Coordenar ações na área de medicamentos e alimentos; Autorizar o
funcionamento de empresas de fabricação, distribuição, importação
e comercialização dos produtos de saúde, conceder registros de
produtos entre outros.
RDC nº200/2017: regulamenta o registro de novos fármacos e
estabelece os critérios para concessão e renovação do registro de
medicamentos com princípios ativos sintéticos e semissintéticos,
classificados como novos, genéricos e similares.
Fonte:https://binged.it/
2Z1IYFO
6.
Público
Vigilância Sanitária
(VISA)
As açõesde vigilância sanitária dirigem-se,
geralmente, ao controle de bens, produtos e serviços que
oferecem riscos à saúde da população, como alimentos,
produtos de limpeza, cosméticos e medicamentos. Realizam
também a fiscalização de serviços de interesse da saúde,
como escolas, hospitais, clubes, academias, parques e
centros comerciais, e ainda inspecionam os processos
produtivos que podem pôr em riscos e causar danos ao
trabalhador e ao meio ambiente.
Público
A Lei nº5.991/1973 define que medicamento é “todo
produto farmacêutico, tecnicamente obtido ou elaborado,
com finalidade profilática, curativa, paliativa ou para fins de
diagnóstico”. Dessa forma, qualquer produto que tenha
objetivos terapêuticos, independente de terem origem
vegetal, animal, mineral ou sintética, deve ser considerado
medicamento e necessita ser registrado.
Lei nº 5.991/1973
12.
Público
A Lei nº13.021 dispõe sobre as novas regras para o
funcionamento das farmácias, regulamentando as ações e serviços
de assistência farmacêutica executados tanto para o âmbito público
quanto para o privado. No âmbito da assistência farmacêutica, as
farmácias de qualquer natureza requerem, obrigatoriamente, para
seu funcionamento, a responsabilidade e a assistência técnica de
farmacêutico habilitado na forma da lei. O único prossional
habilitado para ser responsável técnico para farmácias de qualquer
natureza é o farmacêutico.
Lei nº 13.021/2014
13.
Público
Lei nº 9.787/1999
Essalei é considerada o marco da criação dos
medicamentos genéricos, e por esse motivo é
conhecida como a “Lei dos Genéricos”. Ela de ne os
conceitos de medicamentos de referência, genérico
e similar. A seguir, veja a diferença entre cada um
deles.
14.
Público
RDC 585/2013 dispõesobre as atribuições do farmacêuticas;
RDC 344/1998 Medicamentos Psicotrópicos;
RDC 44/2009 e 471/2021 Medicamentos Antibióticos
RDC 357/2020 trata sobre algumas mudanças na dispensação
de medicamentos controlados devido a pandemia estendida
até maio 2023;
RDC 586/2013 dispõe sobre regulamentação da prescrição
farmacêutica.
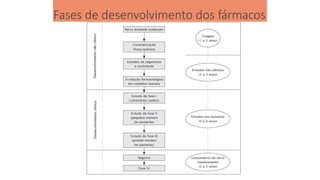
Fases de desenvolvimentodos fármacos
Todo medicamento que vai ser lançado no mercado passa por
diversas etapas de pesquisas e testes para aprovação pelo órgão
competente do país de origem do fabricante (ANVISA-BR);
O processo de regulamentação de um medicamento é longo (10 a 12
anos), rigoroso e custa muito caro (milhões ou bilhões de dólares);
Deve cumprir várias etapas: seleção da molécula, ensaios pré- clínicos
(in vivo/in vitro), testes clínicos (Fases I a IV- humanos);
Comprovar que o produto não provocará reações prejudiciais à vida
das pessoas.
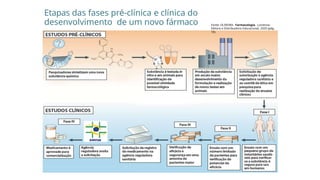
Etapas das fasespré-clínica e clínica do
desenvolvimento de um novo fármaco Fonte: OLIVEIRA. Farmacologia. Londrina:
Editora e Distribuidora Educacional, 2020 (pág.
10).
19.
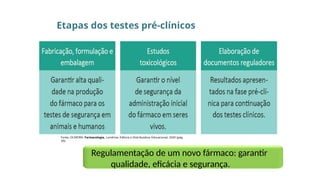
Etapas dos testespré-clínicos
Fonte: OLIVEIRA. Farmacologia. Londrina: Editora e Distribuidora Educacional, 2020 (pág.
20).
Regulamentação de um novo fármaco: garantir
qualidade, eficácia e segurança.
20.
Ensaios toxicológicos dafase pré-clínica
Avaliação inicial da toxicidade da molécula durante a
otimização do protótipo;
Testes de toxicidade: genotoxicidade, farmacologia
cardiovascular, toxicidade de curta duração em animais;
Natureza e mecanismos de possíveis efeitos tóxicos da
molécula;
Efeitos tóxicos inaceitáveis em órgãos-alvo exclusão
da molécula nessa fase do desenvolvimento do
fármaco
Estudos de toxicidade: determinar as condições de segurança
para se iniciar os testes sem seres vivos (animais e humanos);
Individualizados de acordo com o objetivo do
tratamento definido.
21.
E por queainda são feitos testes em animais?
Parte do protocolo para liberação dos testes
em humanos
Pré-requisito para os testes em humanos
Avaliar os efeitos desejados e a toxicidade
Garantir a segurança
22.
Ensaios toxicológicos dafase pré-clínica
Estudos de toxicidade de dose única
(Aguda);
Toxicidade de doses repetidas;
Toxicidade reprodutiva;
Genotoxicidade;
Tolerância local;
Carcinogenicidade;
Avaliação da segurança farmacológica;
Toxicocinética (ADME).
24.
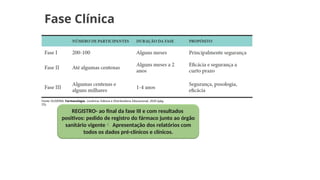
Fase Clínica
Etapafinal para a aprovação de um novo fármaco;
Após atender aos critérios da fase pré-clínica e os ajustes
necessários na molécula, forma farmacêutica e
dosagem o protocolo do estudo clínico deve ser
aprovado pelo Comitê de Ética e Pesquisa e à ANVISA
autorização dos testes clínicos;
1º humanos sadios e após com pessoas que
apresentam a doença a ser tratada;
Realização:
Ensaios clínicos comparados com medicamentos
placebo;
Ensaios clínicos comparados com medicamentos já
existentes no mercado.
25.
Fase Clínica
REGISTRO- aofinal da fase III e com resultados
positivos: pedido de registro do fármaco junto ao órgão
sanitário vigente Apresentação dos relatórios com
todos os dados pré-clínicos e clínicos.
Fonte: OLIVEIRA. Farmacologia. Londrina: Editora e Distribuidora Educacional, 2020 (pág.
25).
26.
Fase IV-
Farmacovigilância
Estudosde vigilância pós-comercialização;
Análise de aparecimento e frequência de novas reações
adversas;
Análise das RAM detectadas possibilita conhecer o perfil de
reações adversas melhor uso do arsenal farmacológico;
Notificação da RAM: é um instrumento fundamental para
alimentar o sistema de farmacovigilância
- Notificação espontânea: relato de uma suspeita de
reações adversas em formulário específico;
- Busca ativa: monitoramento intensivo de possíveis reações
adversas relacionadas a medicamentos;
Eventos adversos detectados podem levar a restrições de uso
em grupos específicos ou até mesmo a retirada do produto do
mercado.
Fonte:https://
binged.it/
27.
Tragédia da Talidomida
Contudo,o marco maior da farmacovigilância foi a tragédia
causada pela talidomida no início da década de 1960. Esse
episódio impulsionou a sistematização dos primeiros esforços
internacionais para abordar questões de segurança no uso de
medicamentos. Naquela época, milhares de crianças
nasceram com malformação congênita (focomelia) em
decorrência da utilização do medicamento talidomida por
suas mães, para o alívio de enjoos da gestação.
28.
Conceitos em farmacovigilância:
EVENTOADVERSO: é um evento desfavorável que ocorre durante ou
após o uso de medicamento ou outra intervenção.
Exemplos: infecções relacionadas à assistência saúde; flebite; obstrução de
sonda para alimentação; reações adversas a medicamentos; eventos
adversos por desvios da qualidade de medicamentos; eventos adversos
decorrentes do uso não aprovado de medicamentos (uso off label);
interações medicamentosas; inefetividade terapêutica total ou parcial;
erros de medicação potenciais e reais, queda do paciente.
29.
Conceitos em farmacovigilância:
REAÇÃOADVERSA A MEDICAMENTO (RAM): é qualquer resposta prejudicial ou
indesejável, não intencional, a um medicamento, que ocorre nas doses
usualmente empregadas no homem para profilaxia, diagnóstico, terapia da
doença ou para a modificação de funções fisiológicas.
EFEITO COLATERAL: é um efeito não pretendido (adverso ou benéfico) causado
por medicamento utilizado em doses terapêuticas. A palavra “colateral” denota
algo de importância secundária.
30.
Patentes dos medicamentos
1883: Convenção de Paris- proteção da propriedade intelectual estabeleceu o
1º acordo internacional (Sistema Internacional da Propriedade Industrial);
Instrumento regulatório para a proteção das invenções de forma internacional;
Lei nº 9279 de 14 de maio de 1996- Lei da propriedade industrial no BR:
prazo de 20 anos de patente para medicamentos a partir da aprovação do
pedido de desenvolvimento de nova molécula;
Após a expiração da patente abre-se a porta para a produção de genéricos.
35.
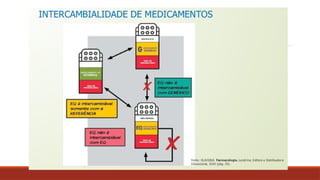
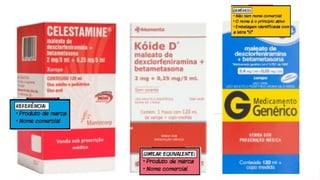
ATIVIDADE INTERCAMBIALIDADE
1) Nareceita médica está prescrito Paracetamol 750mg de 8h/8h. Qual medicamento
posso fazer a intercambialidade?
2) Na receita médica é prescrito Tylenol 750mg de 8h/8h. Qual medicamento posso
fazer a intercambialidade?
3) Na receita médica é prescrito Tylemax Suspensão oral, criança de 11kg, 5ml de
6h/6h. Qual medicamento posso fazer a intercambialidade?
4) Na receita médica está prescrito Bi-Profenid 150mg, 1 comprimido 1x dia. Qual
medicamento posso fazer a intercambialidade?
5) Na receita médica está prescrito Cisteil xarope adulto 40mg/ml, tomar 15ml 1 x dia.
Qual medicamento posso fazer a intercambialidade?
36.
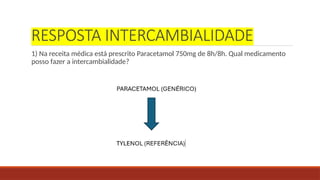
RESPOSTA INTERCAMBIALIDADE
1) Nareceita médica está prescrito Paracetamol 750mg de 8h/8h. Qual medicamento
posso fazer a intercambialidade?
37.
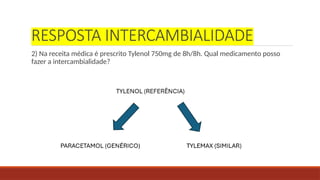
RESPOSTA INTERCAMBIALIDADE
2) Nareceita médica é prescrito Tylenol 750mg de 8h/8h. Qual medicamento posso
fazer a intercambialidade?
38.
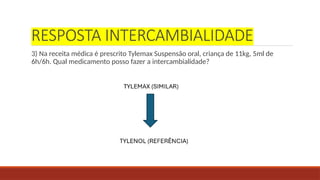
RESPOSTA INTERCAMBIALIDADE
3) Nareceita médica é prescrito Tylemax Suspensão oral, criança de 11kg, 5ml de
6h/6h. Qual medicamento posso fazer a intercambialidade?
39.
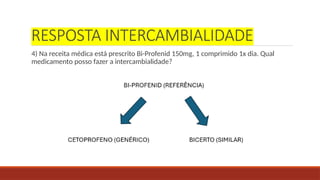
RESPOSTA INTERCAMBIALIDADE
4) Nareceita médica está prescrito Bi-Profenid 150mg, 1 comprimido 1x dia. Qual
medicamento posso fazer a intercambialidade?
40.
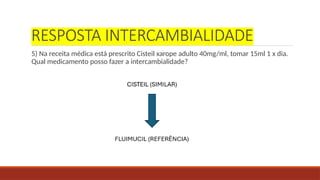
RESPOSTA INTERCAMBIALIDADE
5) Nareceita médica está prescrito Cisteil xarope adulto 40mg/ml, tomar 15ml 1 x dia.
Qual medicamento posso fazer a intercambialidade?
Público
O que sãomedicamentos controlados?
Medicamentos controlados são fármacos que incluem em sua composição
substâncias sujeitas a controle especial conforme listas do Ministério da Saúde.
Essas substâncias agem sobre o sistema nervoso central e podem causar
dependência física ou psíquica, daí a necessidade de controlar sua produção e
comercialização. Para que sejam vendidos em farmácias e drogarias, exigem
prescrição médica em receita de controle especial ou acompanhada por uma
notificação de receita.
São exemplos de remédio controlado alguns analgésicos fortes como a
morfina e medicamentos que ajudam no controle do vício, como a metadona.
Muitos ansiolíticos, tranquilizantes e estimulantes também fazem parte dessa
categoria de remédios.
A legislação completa sobre a produção e comercialização de substâncias e
medicamentos controlados se encontra na Portaria 344 de 1998.
44.
Público
Tipos de medicamentoscontrolados As diferentes classes/categorias dos
medicamentos controlados se referem às substâncias de que eles são feitos e
determinam as restrições específicas a que são submetidos. Nesse sentido, a
Portaria 344/98 divide as substâncias controladas em listas identificadas por
letras que vão de “A” a “F” e apresentam algumas subdivisões.
Prescrição de emergência:
Deveconter os seguintes dados: nome do paciente, nome do
→
medicamento, posologia, diagnóstico ou CID (Código Internacional de
Doenças), justificativa do caráter emergencial do atendimento, data,
número da inscrição no conselho regional e assinatura do prescritor;
Ao dispensar ou aviar o medicamento deve-se anotar a identificação do
→
comprador constando o nome, endereço completo, telefone (se houver) e
número do documento de identidade e órgão expedidor;
Depois de atendida, a receita de emergência deve ser apresentada à
→
autoridade sanitária estadual ou municipal ou distrito federal dentro de 72
horas (3 dias) para visto;
Deve ser arquivada juntamente com as outras receitas de medicamentos
→
controlados.
Tipos de receitas e notificações:
52.
Como avaliar areceita ou notificação?
LISTA DE CHECAGEM P/ AVALIAÇÃO DA RECEITA:
✔ Nome e dados do paciente;
✔ Nome do prescritor, Inscrição no Conselho Regional
(CRM,CRMV,CRO) em carimbo ou na descrição da receita e
assinatura;
✔ Endereço e dados do médico ou clínica atuante;
✔ Classificação de uso interno ou externo (receita);
✔ Nome do medicamento (Referência ou similar) ou Nome genérico;
✔ Concentração/Dosagem;
✔ Quantidade a ser dispensada;
✔ Forma farmacêutica;
✔ Posologia (dose e frequência);
✔ Duração do tratamento;
✔ Está dentro do prazo de validade?
✔ A quantidade prescrita pode ser dispensada?
54.
Como não erraro tipo de
receita no momento da
dispensação?
ATIVIDADE RECEITA ENOTIFICAÇÃO
Avalie abaixo se o medicamento prescrito está conforme legislação, se
não estiver de acordo com a RDC´s 344/98 ou 471/2021 explicar o motivo
da não dispensação.
1) Medicamento zolpidem 10mg, prescrito em receituário comum, com
todos os dados obrigatórios, chegou na farmácia para comprar o
medicamento. Você pode dispensar? Porquê?
2) Medicamento Morfina e Venvanse prescritos na mesma notificação A.
Você pode dispensar? Porquê?
3) Rivotril prescrito na notificação especial B2. Você pode dispensar?
Porquê?
62.
ATIVIDADE RECEITA ENOTIFICAÇÃO
4) Médico prescreveu antibiótico dia 01/10/2024. Paciente foi a
farmácia com a receita dia 12/10/2024, para efetuar a compra.
Baseado nessas informações, você pode dispensar o antibiótico?
5) Médico prescreveu TYLEX dia 22/03/2024. Paciente foi a farmácia
com a receita dia 22/04/2024, para efetuar a compra. Baseado
nessas informações, você pode dispensar esse medicamento?