
Recomendados
Mais conteúdo relacionado
Mais procurados
Mais procurados (20)
Semelhante a Lysosomes mbb
Semelhante a Lysosomes mbb (20)
Último
Último (20)
Lysosomes mbb
- 2. In 1955, Belgian scientist Christian de Duve observed that the cells released an enzyme called acid phosphatase in much larger amounts when they were repeatedly frozen and thawed before centrifugation de Duve C (1975) Exploring cells with a centrifuge. Science 189, 186-194
- 3. To explain this phenomenon, de Duve suggested that the digestive enzyme must have been encased in some sort of membrane-bound organelle within the cell. After estimating the probable size of the lysosomes, he was able to identify the organelle in images produced with an electron microscope
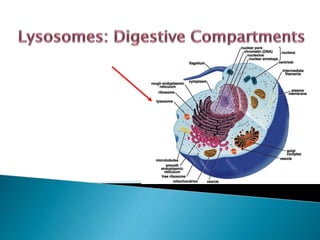
- 4. A lysosome is a membranous sac of digestive enzymes called hydrolytic enzymes that can digest all major classes of macromolecules synthesized by Golgi apparatus only in animal cells. These organelles are simple, spherical membrane bound surrounded by a single membrane. They range in size from 25nm to 1μm in diameter. Lysosomes digest food and worn out cell parts that are no longer used by the cell and are sometimes considered “Suicide Sacs” and plays a role in waste disposal. With the exception of erythrocytes, lysosomes are found in all eukaryotic cell types old organelles go to die! Lysosome = lyso (dissolving), some (body)
- 5. i. Primary lysosomes :contain digestive enzymes ii. Secondary lysosomes: contain digestive enzymes + the digested material. iii. Residual bodies- undigested material inside these bodies iv. Lipofuscin pigments Primary lysosome Phagosome Types of lysosomes and their formation Phagosome Residual BodiesLipofuscin pigments Autophgaic vacoule Residual Bodies
- 6. Lysosomal enzymes can hydrolyze proteins, fats, polysaccharides, and nucleic acids via. action of catabolic enzymes like proteases, lipases, carbohydrases and nucleases respectively. Enzymes present in the lysosomes are synthesized in rough endoplasmic reticulum and are then transported through the cytoplasm by transport vesicles into Golgi apparatus through cis-Golgi or forming face. These enzymes are further processed in Golgi apparatus and then budded off from trans-Golgi or maturing face of Golgi apparatus in the form of primary lysosome.
- 7. They are formed when primary lysosomes fuses with phagocytic vesicles, thus exposing the vesicles contents to lysosomal enzymes. The enzymes present in the primary lysosomes digest the food present and the soluble substances are diffused into the cytoplasm of the cell. Undigested material containing vacuole known as residual body is expelled out by exocytosis. Other forms of Lysosomes: Phagosomes: When the primary lysosome fuses with a specialized white blood cell, the phagocyte, an activated phaogsome or phagocytic vesicles formed. They fight against pathogen by engulfing them very rapidly than the ordinary phagocytes. Autophagic vacuoles: During starvation or after the destruction of cell components especially the liver cells and cells destroyed during metamorphosis fuse with the primary lysosome to form autophagic vacuoles or cytolysosomes.
- 8. A typical lysosome contains at least 50 different hydrolytic enzymes produced in the rough ER and targeted to these organelles. Important enzymes present in the lysosome are acid hydrolases, proteases, lipases and acid phosphatases. These enzymes are capable of digesting organic molecules like lipids, proteins nucleic acids and polysaccharides under acidic conditions. So these are called “Suicidal Bags" They share an important property: all have their optimal activity at an acid pH and thus are acid hydrolases and works best in the acidic (pH=4.6-5) inside the lysosome Lysosomal enzymes trafficked to lysosome via mannose-6-phosphate receptor pathway
- 9. It delivers the degradative enzymes or lysosomal proteins and co-factors to the lysosome through mannose-6-phosphate receptor The process requires the sequential action of two enzymes, phosphotransferase and diesterase. In the Golgi apparatus, mannose-6-phosphate(M-6-P) is covalently linked to soluble enzymes via N-linked oligosaccharides destined for lysosomes. N-linked oligosaccharides helps in targeting lysosomal enzymes to lysosomes and prevent their secretion. Clathrin-coated vesicles transport lysosomal proteins from the trans-Golgi network to maturing endosomes M-6-P is then recognised by M-6-P receptors(M-6-PR) in trans-golgi and delivers them to late endosome
- 10. M-6-P marker separates glycoproteins destined for the lysosome from secretory glycoproteins Failure to acquire this marker results in mistargeting of lysosomal enzymes; they will not enter the lysosome and substrate breakdown will not occur The receptor–protein complex then moves to the late endosome where lower pH causes dissociation M-6-P receptor then retrieved in late endosome and trafficked for re-use in trans-golgi (recognised via C-terminal tail). The hydrolase moves on into the lysosome and the receptor is recycled either to the Golgi to pick up another ligand, or to the plasma membrane. The final steps in the maturation of the lysosomal enzyme include proteolysis, folding and aggregation
- 11. Adaptins bridge the M6P receptor to clathrin. Hydrolases are transported to the late endosome which later matures into a lysosome. Acidic pH causes hydrolase to dissociate from the receptor.
- 12. ENZYME SUBSTRATE Acid phosphatase Phosphate esters Acid ribonuclease RNA Acid deoxyribonuclease DNA Glycosidases Polysaccharides Protease Proteins and peptides Lipase Lipids Phospholipase Phospholipids
- 13. • They hydrolyse proteins, fats, polysaccharides, and nucleic acids. • Can destroy the cell by autodigestion (autophagy). • Can fuse with food vacuoles to digest food, (when a food item is brought into the cell by phagocytosis). • Can also fuse with another organelle or part of the cytosol. This process of autophagy called recycling which renews the cell • They digest unwanted particles • They help white blood cells to destroy bacteria Function of Lysosomal enzymes
- 14. Lysosomal membrane performs important functions: Sequesters potentially destructive hydrolytic enzymes from the cytosol High internal proton concentration is maintained by a proton transporter (H+- ATPase) on the lysosomal membrane (Arai et al.,1993) Maintains the optimal acidic environment for enzyme activity by pumping H+s inward from the cytosol to the lumen Membrane prevents digestive enzymes leaking out and potentially destroying vital cell components
- 15. Lysosomal membranes contain a variety of highly glycosylated integral proteins whose carbohydrate chains are thought to form a protective lining that shields the membrane from attack by the enclosed enzymes. Mannose 6-phosphate residues in lysosomal enzymes act as an “address” for delivery of these proteins to lysosomes Many single-celled organisms ingest food particles, which are then enzymatically disassembled in a lysosome. The resulting nutrients pass through the lysosomal membrane into the cytosol.
- 16. Extracellular (large): phagocytosis (cellular eating) Extracellular (small): ◦ pinocytosis (cellular drinking) ◦ receptor-mediated endocytosis Intracellular: autophagy
- 17. Within the acidic compartments, the endosomes and lysosomes, macromolecules, complex lipids, and oligosaccharides are degraded into their building blocks by hydrolytic enzymes. The resulting catabolites are exported from the lysosome and reused in cellular metabolism (Luzio et al., 2007). This export is mediated by transport proteins present in the limiting lysosomal membrane. These transporters make use of the energy conserved in the proton gradient along the membrane and co-transport small molecules or ions together with protons ◦ e.g. transport proteins for hexoses (GLUT-8) (Schmidt et al., 2009) ◦ Cobalamin, essential for degradation of branched chain amino acids, odd chain fatty acids, and C1-metabolism delivered to the cell via the lysosomes, which illustrates the role of lysosomes for cellular nutrition Other metal ions like iron ions which are essential for many cellular processes from oxygen-transport to respiration are delivered or recycled through the lysosomal compartment (Rutsch et al.,2009).
- 18. Functions of lysosomes The presence within a cell of what is, in essence, a bag of destructive enzymes suggests a number of possible functions. In mammals, phagocytic cells, such as macrophages and neutrophils, function as scavengers that ingest debris and potentially dangerous microorganisms. Ingested bacteria are generally inactivated by the low pH of the lysosome and then digested enzymatically. The macromolecules that are degraded in the lysosome arrive by endocytosis, phagocytosis, or autophagy, receptor-mediated endocytosis. They are involved in intracellular and extracellular digestion since they have enzymes to digest the phagocytosed food particles present in food vacuoles.
- 19. Exo-cytosis: Sometimes enzymes of primary lysosome are released from the cell. This occurs during the replacement of cartilage by bone during development. Similarly the matrix of bone may be broken down during the remodelling of bone that can occur in response to injury, new stresses and so on. Autolysis: It is the self destruction of a cell by release of the contents of lysosomes within the cell. It is normal event in some differentiation processes and may occur throughout a tissue, as when a tadpole tail is reabsorbed during metamorphosis. It also occurs after cells die. Sometimes it occurs as a result of certain lysosomal diseases or after cell damage. Recycling of important constituents: As a result of phagocytosis and digestion of different components, the lysosomes help in the recycling of important components of the cytoplasm
- 20. They help in nutrition of the cell by digesting food, as they are rich in various enzymes which enable them to digest almost all major chemical constituents of the living cell. Help in defence by digesting germs, as in white blood cells. Help in cleaning up the cell by digesting damaged material of the cell. Provide energy during cell starvation by digestion of the cells own parts (autophagic, auto : self; phagos: eat up). Help sperm cells in entering the egg by breaking through (digesting) the egg membrane. In plant cells, mature xylem cells lose all cellular contents by lysosome activity. When cells are old, diseased or injured, lysosomes attack their cell organelles and digest them. In other words lysosomes are autophagic, i.e. self devouring.
- 21. Phagocytosis – cellular process of ingestion, in which the plasma membrane engulfs substances and pinches off to form a particle- containing vacuole Lysosomes may fuse with food-filled vacuoles, and their hydrolytic enzymes digest the food. The food vacuole formed in this way then fuses with a lysosome, whose enzymes digest the food. e.g. Amoebas and many other protists eat by engulfing smaller organisms or other food particlesby the process of phagocytosis Some human cells also carry out phagocytosis. Among them are macrophages, a type of white blood cell that helps defend the body by engulfing and destroying bacteria and other invaders.
- 22. Apoptosis: It is a programmed cell destruction in multi-cellular organisms. This process is important during metamorphosis and development. Lysosomes can be used to kill cells when they are supposed to be destroyed, some cells have to die for proper development in an organism e.g.: when a tadpole becomes a frog, lysosomes digest away the cells of the tail and tail gets re-absorbed ◦ The fingers of a human embryo are at first webbed, but they are free from one another as a result of lysosomal degradation during fetal development. ◦ In case of plants, virus infected plant cell auto-destructs and even cells around it to wall off virus. e.g. Brown spots on leaves ◦ if cell grows improperly this self-destruct mechanism is triggered to remove damaged cell ◦ cancer over-rides this to enable tumor growth There are sensors in the cell that monitor growth. They trigger self-destruct when they sense processes. These all shows Feedback mechanism
- 23. Lysosomes also play a key role in organelle turnover, that is, the regulated destruction of the cell’s own organelles and their replacement Lysosomes also use enzymes to recycle the cell’s own organelles, organic material and macromolecules by the process called autophagy. Lysosomes may engulf other cellular organelles or part of the cytosol and digest them. Resulting monomers are released into the cytosol where they can be recycled into new macromolecules ◦ e.g. A human liver cell, for example, recycles half of its macromolecules each weak Lysosome Lysosomes: Autophagy Peroxisome Mitochondrion Vesicle Digestion
- 24. During this process (autophagy) an organelle, such as the mitochondrion surrounded by a double membrane to produce a structure called an autophagosome. The outer membrane then fuses with a lysosome to produce an autophagolysosome in which the enclosed organelle is degraded and the breakdown products are made available to the cell. If a cell is deprived of nutrients, a marked increase in autophagy is observed. Under these conditions, the cell acquires energy to maintain its life by cannibalizing its own organelles. In recent years, autophagy has also been shown to help protect an organism against intracellular threats ranging from abnormal protein aggregates to invading bacteria.
- 25. If autophagy is blocked in a particular portion of the brain of a laboratory animal, that region of the nervous system experiences massive loss of nerve cells. These findings reveal the importance of autophagy in protecting brain cells from the continuous damage to proteins and organelles that is experienced by these long-lived cells. Once the digestive process in the autophagolysosome has been completed, the organelle is termed a residual body. Depending on the type of cell, the contents of the residual body may be eliminated from the cell by exocytosis, or they may be retained within the cytoplasm indefinitely as a lipofuscin granule. Lipofuscin granules increase in number as an individual becomes older; accumulation is particularly evident in long-lived cells such as neurons, where these granules are considered a major characteristic of the aging process
- 26. The steps involved in the autophagic pathway
- 27. Diseases characterized by the deficiency of a single lysosomal enzyme and the corresponding accumulation of undegraded substrate are called lysosomal storage disorders. Lack of a specific lysosomal enzymes causes substrate accumulation which interferes with lysosomal metabolism and other cellular functions Lysosomal dysfunction has a profound impact on cell homeostasis, resulting in manifold pathological situations, including infectious diseases, neuro- degeneration and aging. They can be classified according to the stored substances, as sphingolipidoses, mucopolysaccharidoses, mucolipidoses, glycoprotein and glycogen storage diseases Most of these diseases result from deficiencies in single lysosomal enzymes
- 28. Tay-Sachs disease: brain impairment by accumulation of lipids. a metabolic disorder involving a missing or inactive lysosomal enzyme in nerve cells. In these cases, the lysosomes fill to capacity with macromolecules that cannot be broken down. The nerve cells become so full of these lysosomes that the child dies. Pompe’s disease: In 1965, H.G. Hers explained the absence of an unimportant lysosomal enzyme, α-glucosidase that breaks down glycogen - glycogen accumulation which damages the liver and could lead to the development a fatal inherited condition known as Pompe’s disease. In the absence of α-glucosidase, undigested glycogen accumulated in lysosomes, causing swelling of the organelles and irreversible damage to the cells and tissues. Niemann-Pick disease: It leads to the accumulation not only of cholesterol, but also of sphingomyelin, glycosphingolipids, sphingosine, and others in multilamellar storage bodies
- 29. Gaucher’s disease: Most common of lysosomal storage diseases. It is a genetic disorder where there is accumulation of lipids in cells and organs. It results from a mutation in the gene that encodes a lysosomal enzyme required for the breakdown of glycolipids. Signs & Symptoms: ◦ Enlarged liver and spleen ◦ Low number of red blood cells (anemia) ◦ bone abnormalities such as bone pain, fractures, and arthritis. ◦ Problems with central nervous system
- 30. LSD lysosomal defects give rise to swollen lysosomes, developmental and degenerative defects with varying involvement of the nervous system due to ‘storage’ of material in the lysosome. LSDs cause death in childhood (generally) after normal infancy LSDs are essentially incurable, but some are treatable to varying degrees Model organisms are helping to define the biology of the LSDs, in particular the ‘pathogenic cascade’