Baixado 10 vezes




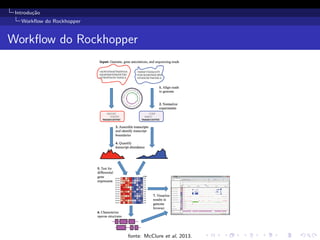


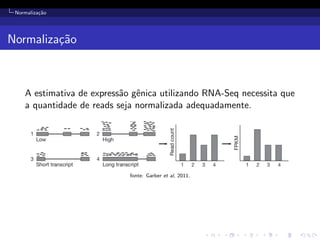



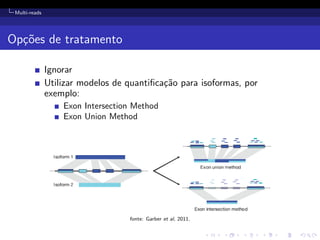








O documento discute o uso da análise de RNA-Seq para quantificar a abundância de transcritos bacterianos. Descreve o workflow típico de análise, incluindo alinhamento de reads, contagem e normalização, e discute fontes de variabilidade e desafios como reads multiplas. O documento também avalia a precisão da estimativa de abundância de transcritos comparando com qRT-PCR e simulações.


































