Prnp
•Transferir como PPTX, PDF•
3 gostaram•1,170 visualizações

talking about PrNp gene and prion disease .

Recomendados
Mais conteúdo relacionado
Mais procurados
Mais procurados (20)
Semelhante a Prnp
Semelhante a Prnp (20)
Último
Último (20)
Prnp
- 1. Prion Protein Bahaaddin Ahmed Saber 27-05-2015
- 2. OUT LINE Definition Gene Expression of Prion protein Function of protein Diagnosis Disease affect by prion protein Treatment And Prevention
- 3. Definition of Prion Prion: A small proteinaceous infectious disease-causing agent that is believed to be the smallest infectious particle. A prion is neither bacterial nor fungal nor viral and contains no genetic material. Prions have been held responsible for a number of degenerative brain diseases, including Mad cow disease, Creutzfeldt-Jakob disease, fatal familial insomnia ,kuru, and an unusual form of hereditary dementia known as Gertsmann-Straeussler-Scheinker disease.
- 4. PRNP gene The PRNP gene is located on the short (p) arm of Chromosome 20 at position 13. More precisely, the PRNP gene is located from base pair 4,686,150 to base pair 4,701,587 on chromosome 20.
- 7. What is chromosome 20? Humans normally have 46 chromosomes in each cell, divided into 23 pairs. Two copies of chromosome 20, one copy inherited from each parent, form one of the pairs. Chromosome 20 spans about 63 million DNA building blocks (base pairs) and represents approximately 2 percent of the total DNA in cells. Identifying genes on each chromosome is an active area of genetic research. Because researchers use different approaches to predict the number of genes on each chromosome, the estimated number of genes varies. Chromosome 20 likely contains 500 to 600 genes that provide instructions for making proteins. These proteins perform a variety of different roles in the body.
- 8. Function of Prion protein • The PRNP gene provides instructions for making a protein called prion protein (PrP), which is active in the brain and several other tissues. Although the precise function of this protein is unknown, researchers have proposed roles in several important processes. These include the transport of copper into cells and protection of brain cells (neurons) from injury (neuroprotection). Studies have also suggested a role for PrP in the formation of synapses, which are the junctions between nerve cells (neurons) where cell-to-cell communication occurs. • Different forms of PrP have been identified. The normal version is often designated PrPC to distinguish it from abnormal forms of the protein, which are generally designated PrPSc.
- 9. What other names do people use for the PRNP gene or gene products? AltPrP … ASCR… CD230 antigen……CJD GSS…. MGC26679… PRIO_HUMAN prion protein (p27-30) (Creutzfeldt-Jakob disease, Gerstmann-Strausler-Scheinker syndrome, fatal familial insomnia) PRIP…PrP PrP27-30 PrP33-35C PrPc PrPSc
- 10. Disease Relation with Prion Protein - Creutzfeldt-Jacob disease (CJD) and variant CJD in humans
- 11. Disease Relation with Prion Protein Scrapie ( In Sheep).
- 12. Disease Relation with Prion Protein Cow Mad disease :
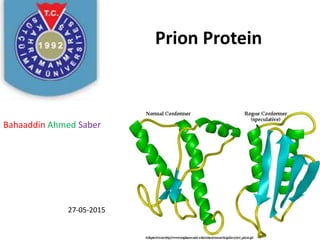
- 13. Models of PrPC to PrPSc conversion. (A) The heterodimer model proposes that upon infection of an appropriate host cell, the incoming PrPSc (orange) starts a catalytic cascade using PrPC (blue) or a partially unfolded intermediate arising from stochastic fluctuations in PrPC conformations as a substrate, converting it by a conformational change into a new β-sheet–rich protein. The newly formed PrPSc (green-orange) will in turn convert new PrPC molecules. (B) The noncatalytic nucleated polymerization model proposes that the conformational change of PrPC into PrPSc is thermodynamically controlled: the conversion of PrPC to PrPSc is a reversible process but at equilibrium strongly favors the conformation of PrPC. Converted PrPSc is established only when it adds onto a fibril-like seed or aggregate of PrPSc. Once a seed is presModels of PrPC to PrPSc conversion. (A) The heterodimer model proposes that upon infection of an appropriate host cell, the incoming PrPSc (orange) starts a catalytic cascade using PrPC (blue) or a partially unfolded intermediate arising from stochastic fluctuations in PrPC conformations as a substrate, converting it by a conformational change into a new β-sheet–rich protein. The newly formed PrPSc (green-orange) will in turn convert new PrPC molecules. (B) The noncatalytic nucleated polymerization model proposes that the conformational change of PrPC into PrPSc is thermodynamically controlled: the conversion of PrPC to PrPSc is a reversible process but at equilibrium strongly favors the conformation of PrPC. Converted PrPSc is established only when it adds onto a fibril-like seed or aggregate of PrPSc. Once a seed is present, further monomer addition is acceleratedent, further monomer addition is accelerated. .
- 14. Diagnosis • MRI scans of the brain • Samples of fluid from the spinal cord (spinal tap) • Electroencephalogram, which analyzes brain waves; this painless test requires placing electrodes on the scalp • Blood tests • Neurologic and visual examinations to evaluate for nerve damage and vision loss
- 15. Diagnosis DIAGNOSING TSEs It can be very difficult to test for PrPSc because it may be present in small amounts and separating it out from PrPC is not trivial. The methods currently being used test dead animal tissue. There are proposed ways to test live animals, such as detecting Prions in blood, urine or Cerebrospinal Fluid (CSI), but these methods are not yet in widespread use. More information about the differences between the conformations of the proteins is needed before these other tests can become a reality. 1Bioassay The most conclusive form of testing is the bioassay. This test works by taking a tissue sample of a suspected infected animal and putting it into a mouse or other animal and waiting for the disease to develop in the model organism. Obviously, this is a very slow and labor-intensive method of testing for the disease. A faster form of testing, but still labor intensive, is Immunohestochemistry. In this method, antibodies that recognize PrPSc are injected into brain tissue, and then observed on slides under a microscope for the presence of these antibodies . If the antibodies are present, then they have attached to infectious prions and the tissue is infectious. Because each slide must be examined under a microscope, this type of test can take time and energy that makes it hard for mass testing. 2Immunoassay Another type of test, the immunoassay, is fast and easier to complete, but only applicable when there are high levels of infectious prions; subsequently, lower levels may go undetected. The Immunoassay works by first adding Protease to brain tissue, which will break down all the non-infectious form of prions and leave only infectious prions. Then, antibodies sensitive to prions are added to the solution. The antibodies used are tagged with a visual marker so that the presence of the prions with show up. Testers can also run the solution on a Gel and the presence bands will mean that there is infectious prion protein. The reason this only works for high levels of infectious protein is that although many PrPScs are resistant to breakdown by proteases, many are not, especially in the early stages of the disease (it is not known why this is the case). Therefore, after proteases have been applied to the mixture, all of the PrPC is broken down, and some of the PrPSc is broken down as well, so only a little bit remains. Furthermore, it is hard to find antibodies that will only bind to the infectious form when we do not know the exact conformation differences, so proteases are a necessary step before adding antibodies.
- 16. Diagnosis Conformation-Dependent Immunoassay 3- Conformation dependent Immunoassay (CDI)Finally, a newer method that is both quick and accurate for low levels of infectious protein. CDI uses tissue taken from a live animal mixed with a chemical that separates infectious from non-infections prions based on their conformations. Next, an antibody tagged with flourescence is added to the separated area, and if the tissue same contains infectious protein, the antibody will fluoresce . 4- Brain Imaging is another diagnostic tool that can be used for humans, but is not applicable to diagnosing animals that may be fed to humans. Furthermore, brain scans are expensive, and would probably only be used if someone is showing symptoms of aTSE, which often means the disease is very advanced. The development of a wider diagnostic test that could be applied to anyone who might be at risk of a TSE would be incredibly valuable because it might be easier to slow down in an early stage of the disease (Committee on Transmissible Spongiform Encephalopathies, 2004).
- 17. 5- Protein Misfolding Cyclic Amplification (PMCA) One of the obstacles to creating a test that does not require brain tissue, as all of the above tests do, is the low level of prions in other tissues. Formulating a blood or urine test would require a means of amplifying the infectious protein to levels that are detectable. This is difficult because since it is only protein, polymerase chain reaction (a very efficient method for amplifying nucleic acids) is not applicable. However, there is a method being developed that may allow an amplification process that would make a blood test more feasible. This amplification process is called Protein Misfolding Cyclic Amplification (PMCA). It has been shown to work experimentally by mixing infected prions with normal prions. The idea behind it is that infectious prions transform normal prions into infectious prions, so the normal prions are there for the infectious to work on changing. However, often the infectious prions form clumps, so they less actively change the normal prion conformations. Therefore, this method uses sonication-pulses of sound waves-to break up infectious prion clumps so that they will spread throughout the tissue mix and transform the normal prions. This seems to work as a method of amplifying infectious protein. Another possible way to get around the low levels of infectious protein is to detect a "surrogate marker" instead of the protein itself. There may be other proteins or molecules that indicate the presence of the infectious prion protein that are easier to detect, and could therefore act as a "surrogate marker" because they, instead of prions, could be tested for in tissue. Another obstacle is how to distinguish among the different strains of TSEs. Knowing this could be integral to determining the source of the disease (inherited, spontaneous, transmitted) and to inventing with more targeted ways of dealing with the different strains disease (Committee on Transmissible Spongiform Encephalopathies, 2004).
- 18. protein misfolding cyclic amplification machine
- 20. Symptoms of Prion disease • Rapidly developing dementia • Difficulty walking and changes in gait • Hallucinations • Muscle stiffness • Confusion • Fatigue • Difficulty speaking
- 21. Treatment • Prion diseases can't be cured, but certain medications may help slow their progress. Medical management focuses on keeping people with these diseases as safe and comfortable as possible despite progressive and debilitating symptoms. 1- Quinacrine 2-Pentosan polysuphate (PPS) 3-Tetracyclic Compounds 4-Flupirtine 5-Potential Treatments (Immunotherapy)
- 22. Thanks for your Attention