
Recomendados
Recomendados
Mais conteúdo relacionado
Mais procurados
Mais procurados (20)
Semelhante a RNA Seq Data Analysis
Semelhante a RNA Seq Data Analysis (20)
Mais de Ravi Gandham
Último
Último (20)
RNA Seq Data Analysis
- 1. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Overview of the previous lecture
- 2. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Overview of the previous lecture
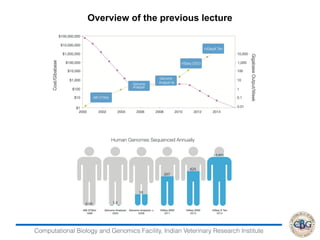
- 3. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Instrument Needed Coverage Throughput/ Run Human Genome/run Cost per Run** Total Cost $/Genome HiSeq 2500 30x 1Tb 11 29.000 29,000 2,536 HiSeq x 10 30x 1 STb 16 15.700 12,000 950 Pacbio RSII 54x 1 Gb 0.000 212 34,374 34,374 PacBio Sequel 50x 5-10 Gb 0.06 700 10,300 10,500 Current Loading Technologies Platform Average Reeds length Advantages Limitation Material Recommended Illumina MiSeq 2 x 300 bps Accurate Short Length 100-200 ng Illumina Mole- culo 5 Kbps Accurate Coverage may Fluctuate 10 ug Pacific Bioscience 15 Kbps Long reads Relatively expensive 10-100 ug Oxford Nanopore 5 Kbps Low ownership cost High error rate 1 ug Long-Range Sequencers Comparison
- 4. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Basics of RNA - seq data analysis
- 5. Computational Biology and Genomics Facility, Indian Veterinary Research Institute • Sequence DNA • De novo sequencing • Reference based re-sequencing - SNP, CNV and indels • Metagenomics - Identify who is there in a mixture of microbes • Sequence RNA • RNA-Seq (Transcriptome wide sequencing) • miRNA - Seq novo sequencing • Novel NcRNAs • Study Protein-DNA/RNA interactions • ChIP-Seq (TFs) • CLIP - Seq (For RNA binding proteins) • Epigenetics • DNA methylation • Histone modification (ChIP-seq) • Nucleosome positioning • Chromosome looping Applications of NGS
- 6. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Certain important key words to be remembered from the previous chapter • Sequencing • both DNA and RNA (with modified protocol) • Short reads • 35, 50, 75, and 100-bp (Solexa and SOLiD) • 400-bp (454) • Ultra-high throughput • 1 to 1.5 billion reads (Solexa and SOLiD) • 2- 4 million reads (454)
- 7. Computational Biology and Genomics Facility, Indian Veterinary Research Institute • The transcriptome : Complete set of transcripts in a cell, both in terms of type and quantity. • Transcriptome analysis - • in understanding the pattern of gene expression to address basic biological questions. • greater insights into biological pathways and molecular mechanisms that regulate cell fate, development, and disease progression. What is a transcriptome? Transcriptome can be studied through RNA-seq/microarray
- 8. Computational Biology and Genomics Facility, Indian Veterinary Research Institute RNA sequencing
- 9. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Aim of RNA-seq experiment • To quantify RNA abundance • To determine the transcriptional structure of genes: start sites, 5’ and 3’ ends, splicing patterns • To quantify the changing expression levels of each transcript during development and under different conditions • To identify variants on the transcripts
- 10. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Basic work flow of Next generation sequencing Wang et al., 2009
- 11. Computational Biology and Genomics Facility, Indian Veterinary Research Institute What can we get out of RNA-seq experiment? Gene expression Alternative splicing Transcript variation Non-coding RNAs RNA -seq Different expression Non- syn SNPs Synonymous SNPs SNPs in 3’- UTRs Allele specific expression Protein changes RNA binding proteins microRNA binding sites
- 12. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Microarray Normal RNA Cy3 Labelling Reverse transcription Experimental RNA Cy5 labelling Hybridize Wash & Scan
- 13. Computational Biology and Genomics Facility, Indian Veterinary Research Institute RNA - Seq vs Microarray “RNA- seq…. is expected to revolutionise the manner in which eukaryotic transcriptome are analysed” Wang et al. Nat Rev Gen, 2009
- 14. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Limitations of microarray platforms • High background levels owing to cross-hybridization; • Limited dynamic range of detection owing to both background and saturation of signals. • Reliance upon existing knowledge about genome sequence;
- 15. Computational Biology and Genomics Facility, Indian Veterinary Research Institute RNA - seq has higher dynamic range For microarrays: • Low signal end: high background noise • High signal end: signals will be saturated With proper depth, NGS can solve this problem well
- 16. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Comparison of Microarray with RNA - seq
- 17. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Experimental considerations
- 18. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Sample preparation • Amount requirement • 100 ug total RNA (several Wold’s studies) • 2 ug total RNA (Center for Medical Genomics, SOLiD) • 50 ng total RNA (collaboration with Pourmand, SOLiD) • Single cell (Tang et al. Nat. Methods, 2009) (when RNA is limiting, approaches to amplify small quantities of RNA exist) • rRNA removal • rRNAs are highly abundant (>90% of total RNA) Solutions: • rRNA depletion kits • Poly-A selection • Using enzymes that selectively degrade uncapped RNA
- 19. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Sample preparation • Reverse transcription (RNA -> cDNA) • oligo dT • Pros: Focusing on polyA ed transcriptions cleaner. • Cons: Bias towards the 3’-end of transcripts • Random primers • Pros: Equal coverage, can be used to study non-polyAed transcripts • Cons: higher proportion of rRNA
- 20. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Single end reads vs paired ends reads 3' 5' 5' 3' 5' 3' 5' 3' AAAA AAAA 3' 5'TTTT cDNA fragments and adapter ligation cDNA conversion R1 R2 5' 3' 3' 5' Sequencing of each fragment R1 will run in the same direction of the reference R2 will run in the opposite direction of the reference
- 21. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Statistical considerations Number of conditions? • Dynamics? • Dose-effect? • Tissue-specificity? Number of replicates? Depth of sequencing? • Biological variation, … • Statistical power, … • Gene expression • Alternative splicing • Allele specific expression ₹
- 22. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Basics of RNA - seq data analysis - II Data processing
- 23. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Data analysis workflow Primary analysis Tertiary analysis • Image acquisition / semiconductor based detection • Base calling and • Quality metrics Secondary analysis • Sequence alignment • Sequence Stats • Consensus Calling • Sequence assembly • Application specific analysis • DNA-Seq • Re-Sequencing • De-novo Sequencing • ChiP Sequencing • RNA-Seq • Epigenetics Data analysis workflow
- 24. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Sequence alignment
- 25. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Sequence alignment ▪ Sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity ▪ Aligned sequences of nucleotide or amino acid residues are typically represented as rows within a matrix. ▪ Gaps are inserted between the residues so that identical or similar characters are aligned in successive columns. 1. To find whether two (or more) genes or proteins are evolutionarily related to each other 2. To observe patterns of conservation (or variability). 3. To find structurally or functionally similar regions within proteins i.e to find the common motifs present in both sequences. 4. To find out which sequences from the database are similar to the sequence at hand Purpose of sequence alignment
- 26. Computational Biology and Genomics Facility, Indian Veterinary Research Institute 1. They are often used interchangeably, they have quite different meanings. 2. Sequence identity refers to the occurrence of exactly the same nucleotide or amino acid in the same position in aligned sequences. 3. The term ‘sequence homology’ is the most important (and the most abused) of the three. • When we say that sequence A has high homology to sequence B, then we are making two distinct claims: • not only are we saying that sequences A and B look much the same, but also that all of their ancestors also looked the same, going all the way back to a common ancestor. Identity vs Similarity vs Homology
- 27. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Sequence Identity Sequence similarity Sequence homology Definition Proportion of identical residues between two sequences. Proportion of similar residues between two sequences. Two residues are similar if their substitution cost is higher than 0. Sequences derived from a common ancestor Expressed as % identity % Similarity Yes or No Rule-of-thumb: If two sequences are more than 100 amino acids long (or 100 nucleotides long) they are considered homologues if 25% of the amino acids are identical (70% of nucleotide for DNA). Twilight zone = protein sequence similarity between ~0-20%
- 28. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Global alignment • assumes that the two sequences are basically similar over the entire length of one another. • forces to match the sequences from end to end, even though parts of the alignment are not very convincingly matching. • most suitable when the two sequences are of similar length and are with a significant degree of similarity throughout. . Computational approaches • Global alignment • Local alignment
- 29. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Local alignment • Identifies segments of the two sequences that match well with no attempt to force the entire sequences into alignment • Parts that appear to have good similarity, according to some criterion are aligned. • Suitable when comparing substantially different sequences, which possibly differ significantly in length, and have only short patches of similarity
- 30. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Give two sequences we need a number to associate with each possible alignment (i.e. the alignment score = goodness of alignment). The scoring scheme is a set of rules which assigns the alignment score to any given alignment of two sequences. • The scoring scheme is residue based: it consists of residue substitution scores (i.e. score for each possible residue alignment), plus penalties for gaps. • The alignment score is the sum of substitution scores and gap penalties. Substitution scores are given by : For DNA : Substitution Matrix for DNA (Purine/Purine or purine/pyramidine substitutions) For proteins : Substitution matrix based on Polarity, Size, Charge or Hydrophobicity Evolutionary distance matrices :- PAM and BLOSUM for protein sequences
- 31. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Types of alignment Pairwise alignment Multiple Sequence alignment Can be Global or Local
- 32. Computational Biology and Genomics Facility, Indian Veterinary Research Institute • Dot Matrix method • The dynamic programming method • Needleman and Wunch • Smith and Watermann • Heuristic methods - FASTA ; BLAST Methods of pairwise alignment • It is a visual graphical representation of similarities between two sequences. • Each axis represents one of the two sequences to be compared. • In the dot matrix method when two sequences are similar over their entire length a line will extend from one corner of the dot plot to the diagonally opposite corner. • If two sequences share only patches of similarity then it will be revealed by diagonal stretches. Dot Matrix method
- 33. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Interpretation of Dot Matrix • Regions of similarity appear as diagonal runs of dots. • Reverse diagonals (perpendicular to diagonal) indicate inversions. • Reverse crossing diagonals (Xs) indicate palindromes. Limitation:- • The dot matrix computer programs do not show an actual alignment.
- 34. Computational Biology and Genomics Facility, Indian Veterinary Research Institute The dynamic programming • Dynamic programming reduces the massive number of possibilities that need to be considered in aligning sequences. • This method was first used for global alignment of sequences by Needleman-Wunch algorithm (1970) and for local alignment by Smith - Waterman algorithm (1981). • Both the algorithms involve initialization, matrix filling (scoring) and trace back steps. The algorithms use either PAM or BLOSUM matrices in the scoring step to fill the score matrix.
- 35. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Global alignment
- 36. Computational Biology and Genomics Facility, Indian Veterinary Research Institute The three main steps in this algorithm are : 1. Initialization 2. Matrix filling 3. Traceback for alignment Initialization 1. Place the two sequences one across the row and other down the column 2. The first column and first row should be a gap 3. Add the cumulative gap cost across the row and other down the column to fill the first column and first row Global alignment (Needleman and Wunch)
- 37. Computational Biology and Genomics Facility, Indian Veterinary Research Institute • Matrix filling • Rules :- • Check the side, top and diagonal values of the box • Box Beside - (add gap cost) • Box top - ( add gap cost) • Diagonal box - (match/mismatch) • Put the highest value in the respective boxes • Proceed to the end of the scoring matrix • Trace back • Start from the end of the matrix and reach the start by tracing back the value obtained in the box • if diagonal - Place the characters • if vertical or horizontal - place a gap in the sequence being pointed by the arrow
- 38. Computational Biology and Genomics Facility, Indian Veterinary Research Institute gap T G A gap 0 -2 -4 -6 A -2 -1 -4 -1 -4 -3 -6 -3 -3 -3 -8 -3 -5 T -4 -1 -3 -1 -6 -2 -5 -2 -3 -4 -5 -4 -4 G -6 -5 -3 -3 -8 0 -4 0 -5 -3 -6 -2 -2 C -8 -7 -5 -5 -10 -4 -2 -2 -7 -1 -4 -1 -4 Matrix Filling - Gap -2; Mismatch -1; Match +1 Box Beside : +gap; Box Top : +gap; Diagonal box : match or mismatch
- 39. Computational Biology and Genomics Facility, Indian Veterinary Research Institute gap T G A gap 0 -2 -4 -6 A -2 -1 -4 -1 -4 -3 -6 -3 -3 -3 -8 -3 -5 T -4 -1 -3 -1 -6 -2 -5 -2 -3 -4 -5 -4 -4 G -6 -5 -3 -3 -8 0 -4 0 -5 -3 -3 -2 -2 C -8 -7 -5 -5 -10 -4 -2 -2 -7 -1 -4 -1 -3 Trace back
- 40. Computational Biology and Genomics Facility, Indian Veterinary Research Institute ATGC - TGA -2+1+1-1 = -1
- 41. Computational Biology and Genomics Facility, Indian Veterinary Research Institute gap G A C T A C gap 0 -1 -2 -3 -4 -5 -6 A -1 0 -2 0 -2 0 -3 0 -1 -2 -4 -1 -1 -3 -5 -2 -2 -3 -6 -3 -3 -5 -7 -4 -4 C -2 -1 -1 -1 -3 0 -1 0 -2 +1 -2 +1 -1 -1 -3 0 0 -2 -4 -1 -1 -3 -5 -2 -2 G -3 -1 -2 -1 -4 -1 -1 -1 -2 0 0 0 -2 +1 -1 +1 -1 0 -2 0 0 -1 -3 -1 -1 C -4 -3 -2 -2 -5 -1 -2 -1 -3 0 -1 0 -2 0 0 0 -1 +1 -1 +1 -1 +1 -2 +1 0 Matrix Filling - Gap -1; Mismatch - 0; Match +1
- 42. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Matrix Filling - Gap -1; Mismatch - 0; Match +1 gap G A C T A C gap 0 -1 -2 -3 -4 -5 -6 A -1 0 -2 0 -2 0 -3 0 -1 -2 -4 -1 -1 -3 -5 -2 -2 -3 -6 -3 -3 -5 -7 -4 -4 C -2 -1 -1 -1 -3 0 -1 0 -2 +1 -2 +1 -1 -1 -3 0 0 -2 -4 -1 -1 -3 -5 -2 -2 G -3 -1 -2 -1 -4 -1 -1 -1 -2 0 0 0 -2 +1 -1 +1 -1 0 -2 0 0 -1 -3 -1 -1 C -4 -3 -2 -2 -5 -1 -2 -1 -3 0 -1 0 -2 0 0 0 -1 +1 -1 +1 -1 +1 -2 +1 0
- 43. Computational Biology and Genomics Facility, Indian Veterinary Research Institute ACG-C GACTAC -1+1+1+0-1+1 = +1 AC-GC GACTAC -1+1+1-1+0+1 = +1
- 44. Computational Biology and Genomics Facility, Indian Veterinary Research Institute gap T G A gap 0 -2 -4 -6 A -2 T -4 G -6 C -8 Matrix Filling - Gap -2; Mismatch -1; Match +1 Box Beside : +gap; Box Bottom : +gap; Diagonal box : match or mismatch
- 45. Computational Biology and Genomics Facility, Indian Veterinary Research Institute gap G A C T A C gap 0 A -2 C -4 G -6 C -8 Matrix Filling - Gap -2; Mismatch - -1; Match +1
- 46. Computational Biology and Genomics Facility, Indian Veterinary Research Institute gap G A C T A C gap A C G C Matrix Filling - Gap -1; Mismatch - 0; Match +1
- 47. Computational Biology and Genomics Facility, Indian Veterinary Research Institute gap T G A gap A T G C Matrix Filling - Gap -2; Mismatch -1; Match +1 Box Beside : +gap; Box Bottom : +gap; Diagonal box : match or mismatch
- 48. Computational Biology and Genomics Facility, Indian Veterinary Research Institute The three main steps in this algorithm are : 1. Initialization 2. Matrix filling 3. Traceback for alignment Initialization 1. Place the two sequences one across the row and other down the column 2. The first column and first row should be a gap 3. Place zeros in first column and first row Matrix filling 1. The value of each box thereon depends on the top, diagonal and side boxes (Box Beside - (add gap cost); Box top - ( add gap cost); Diagonal box - (match/mismatch) 2. If the value is negative - put the value as zero 3. The highest of the three values is placed in the box 4. The same is continued till the end of the matrix Smith and Waterman algorithm (Local alignment)
- 49. Computational Biology and Genomics Facility, Indian Veterinary Research Institute gap T G A gap 0 0 0 0 A 0 0 0 0 0 0 0 0 0 +1 0 +1 0 T 0 +1 0 +1 0 0 0 0 0 0 0 0 0 G 0 0 0 0 0 +2 0 +2 0 0 0 0 0 C 0 0 0 0 0 0 0 0 0 +1 0 +1 0 Matrix Filling - Gap -2; Mismatch -1; Match +1
- 50. Computational Biology and Genomics Facility, Indian Veterinary Research Institute TG TG +1+1=+2
- 51. Computational Biology and Genomics Facility, Indian Veterinary Research Institute gap G C C T A C C C G A A T gap 0 0 0 0 0 0 0 0 0 0 0 0 0 G 0 +1 0 +1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 -2 0 -2 0 -2 0 -2 +1 0 +1 -2 0 0 0 0 0 0 0 0 0 0 0 0 A 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 +1 0 +1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 +2 0 +2 0 +1 0 +1 0 0 0 0 0 A 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 +1 0 +1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 +1 0 +1 0 +3 0 +3 0 0 0 +1 +1 T 0 0 0 0 0 0 0 0 0 0 0 0 0 +1 0 +1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 +1 +1 0 +4 0 +4 0 Matrix Filling - Gap -2; Mismatch -1; Match +1
- 52. Computational Biology and Genomics Facility, Indian Veterinary Research Institute GAAT GAAT 1+1+1+1=+4
- 53. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Global alignment - example
- 54. Computational Biology and Genomics Facility, Indian Veterinary Research Institute gap A T G G C G T gap 0 -2 -4 -6 -8 -10 -12 -14 A -2 +1 -4 +1 -4 -3 -6 -1 -1 -5 -8 -3 -3 -7 -10 -5 -5 -9 -12 -7 -7 -11 -14 -9 -9 -13 -16 -11 -11 T -4 -3 -1 -1 -6 +2 -3 +2 -3 -2 -5 0 0 -4 -7 -2 -2 -6 -9 -4 -4 -8 -11 -6 -6 -8 -13 -8 -8 G -6 -5 -3 -3 -8 -2 0 0 -5 +3 -2 +3 -2 +1 -4 +1 +1 -3 -6 -1 -1 -3 -8 -3 -3 -7 -10 -5 -5 A -8 -5 -5 -5 -10 -4 -2 -2 -7 -1 +1 +1 -4 +2 -1 +2 -1 0 -3 0 0 -2 -5 -2 -2 -4 -7 -4 -4 G -10 -9 -7 -7 -12 -6 -4 -4 -9 -1 -1 -1 -6 +2 0 +2 -3 +1 -2 +1 0 +1 -4 +1 -1 -3 -6 -1 -1 T -12 -11 -9 -9 -14 -6 -6 -6 -11 -5 -3 -3 -8 -2 0 0 -5 +1 -1 +1 -2 0 -1 0 -1 +2 -3 +2 -2 Matrix Filling - Gap -2; Mismatch -1; Match +1
- 55. Computational Biology and Genomics Facility, Indian Veterinary Research Institute gap A T G G C G T gap 0 -2 -4 -6 -8 -10 -12 -14 A -2 +1 -4 +1 -4 -3 -6 -1 -1 -5 -8 -3 -3 -7 -10 -5 -5 -9 -12 -7 -7 -11 -14 -9 -9 -13 -16 -11 -11 T -4 -3 -1 -1 -6 +2 -3 +2 -3 -2 -5 0 0 -4 -7 -2 -2 -6 -9 -4 -4 -8 -11 -6 -6 -8 -13 -8 -8 G -6 -5 -3 -3 -8 -2 0 0 -5 +3 -2 +3 -2 +1 -4 +1 +1 -3 -6 -1 -1 -3 -8 -3 -3 -7 -10 -5 -5 A -8 -5 -5 -5 -10 -4 -2 -2 -7 -1 +1 +1 -4 +2 -1 +2 -1 0 -3 0 0 -2 -5 -2 -2 -4 -7 -4 -4 G -10 -9 -7 -7 -12 -6 -4 -4 -9 -1 -1 -1 -6 +2 0 +2 -3 +1 -2 +1 0 +1 -4 +1 -1 -3 -6 -1 -1 T -12 -11 -9 -9 -14 -6 -6 -6 -11 -5 -3 -3 -8 -2 0 0 -5 +1 -1 +1 -2 0 -1 0 -1 +2 -3 +2 -2 Matrix Filling - Gap -2; Mismatch -1; Match +1
- 56. Computational Biology and Genomics Facility, Indian Veterinary Research Institute gap A T G G C G T gap 0 -2 -4 -6 -8 -10 -12 -14 A -2 +1 -4 +1 -4 -3 -6 -1 -1 -5 -8 -3 -3 -7 -10 -5 -5 -9 -12 -7 -7 -11 -14 -9 -9 -13 -16 -11 -11 T -4 -3 -1 -1 -6 +2 -3 +2 -3 -2 -5 0 0 -4 -7 -2 -2 -6 -9 -4 -4 -8 -11 -6 -6 -8 -13 -8 -8 G -6 -5 -3 -3 -8 -2 0 0 -5 +3 -2 +3 -2 +1 -4 +1 +1 -3 -6 -1 -1 -3 -8 -3 -3 -7 -10 -5 -5 A -8 -5 -5 -5 -10 -4 -2 -2 -7 -1 +1 +1 -4 +2 -1 +2 -1 0 -3 0 0 -2 -5 -2 -2 -4 -7 -4 -4 G -10 -9 -7 -7 -12 -6 -4 -4 -9 -1 -1 -1 -6 +2 0 +2 -3 +1 -2 +1 0 +1 -4 +1 -1 -3 -6 -1 -1 T -12 -11 -9 -9 -14 -6 -6 -6 -11 -5 -3 -3 -8 -2 0 0 -5 +1 -1 +1 -2 0 -1 0 -1 +2 -3 +2 -2 Matrix Filling - Gap -2; Mismatch -1; Match +1
- 57. Computational Biology and Genomics Facility, Indian Veterinary Research Institute gap A T G G C G T gap 0 -2 -4 -6 -8 -10 -12 -14 A -2 +1 -4 +1 -4 -3 -6 -1 -1 -5 -8 -3 -3 -7 -10 -5 -5 -9 -12 -7 -7 -11 -14 -9 -9 -13 -16 -11 -11 T -4 -3 -1 -1 -6 +2 -3 +2 -3 -2 -5 0 0 -4 -7 -2 -2 -6 -9 -4 -4 -8 -11 -6 -6 -8 -13 -8 -8 G -6 -5 -3 -3 -8 -2 0 0 -5 +3 -2 +3 -2 +1 -4 +1 +1 -3 -6 -1 -1 -3 -8 -3 -3 -7 -10 -5 -5 A -8 -5 -5 -5 -10 -4 -2 -2 -7 -1 +1 +1 -4 +2 -1 +2 -1 0 -3 0 0 -2 -5 -2 -2 -4 -7 -4 -4 G -10 -9 -7 -7 -12 -6 -4 -4 -9 -1 -1 -1 -6 +2 0 +2 -3 +1 -2 +1 0 +1 -4 +1 -1 -3 -6 -1 -1 T -12 -11 -9 -9 -14 -6 -6 -6 -11 -5 -3 -3 -8 -2 0 0 -5 +1 -1 +1 -2 0 -1 0 -1 +2 -3 +2 -2 Matrix Filling - Gap -2; Mismatch -1; Match +1
- 58. Computational Biology and Genomics Facility, Indian Veterinary Research Institute ATGGCGT AT - GAGT 1+1-2+1-1+1+1 = +2
- 59. Computational Biology and Genomics Facility, Indian Veterinary Research Institute ATGGCGT ATGA - GT 1+1+1-1-2+1+1 = +2
- 60. Computational Biology and Genomics Facility, Indian Veterinary Research Institute ATGGCGT ATG - AGT 1+1+1-2-1+1+1 = +2
- 61. Computational Biology and Genomics Facility, Indian Veterinary Research Institute ATGGCGT ATG - AGT 1+1+1-2-1+1+1 = +2
- 62. Computational Biology and Genomics Facility, Indian Veterinary Research Institute ATGGCGT AT - GAGT 1+1-2+1-1+1+1 = +2 ATGGCGT ATGA - GT 1+1+1-1-2+1+1 = +2 ATGGCGT ATG - AGT 1+1+1-2-1+1+1 = +2
- 63. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Local alignment
- 64. Computational Biology and Genomics Facility, Indian Veterinary Research Institute gap A T G G C G T gap 0 0 0 0 0 0 0 0 A 0 +1 0 +1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 T 0 0 0 0 0 +2 0 +2 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 +1 0 +1 0 G 0 0 0 0 0 0 0 0 0 +3 0 +3 0 +1 0 +1 0 0 0 0 0 +1 0 +1 0 0 0 0 0 A 0 +1 0 +1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 G 0 0 0 0 0 0 0 0 0 +1 0 +1 0 +1 0 +1 0 0 0 0 0 +1 0 +1 0 0 0 0 0 T 0 0 0 0 0 +1 0 +1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 +2 0 +2 0 Matrix Filling - Gap -2; Mismatch -1; Match +1
- 65. Computational Biology and Genomics Facility, Indian Veterinary Research Institute ATG ATG 1+1+1= +3
- 66. Computational Biology and Genomics Facility, Indian Veterinary Research Institute gap A T G G C G T gap 0 -2 -4 -6 -8 -10 -12 -14 A -2 T -4 G -6 A -8 G -10 T -12 Matrix Filling - Gap -2; Mismatch -1; Match +1
- 67. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Note :- Always take the value of gap cost or mismatch cost a negative value and the values have to be different
- 68. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Drawback of Dynamic programming - Very slow Need for - faster alignment strategies Fast Sequence alignment strategies • Using hash table based indexing - seed extend paradigm, space allowance • Using suffix/prefix tree based - Suffix array, Burrows wheeler transformation and FM index • Merge sorting Strategy: making a dictionary (index) – An example of 4-nt index AAAA: 235, 783, 10083,...... AAAC: 132, 236, 832, 932, ... TTTT: 327, 1328, 5523,...... Algorithms Hashing reads - Eland, MAQ, Mosaik... Hashing reference genome - BFAST, Mosaik, SOAP, ... Hash table based indexing
- 69. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Burrows wheeler transformation and FM index
- 70. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Criteria for choosing an aligner • Global or local • Aligning short sequences to long sequences such as short reads to a reference • Aligning long sequences to long sequences such as long reads or contigs to a reference • Handles small gaps (insertions and deletions) • Handles large gaps (introns) • Handles split alignments (chimera) • Speed and ease of use
- 71. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Short read aligner Aligner Purpose Bowtie Fast BWA small gaps (indels) GSNAP Large gaps (introns) Bowtie 2 Takes care of gaps Long sequence aligner Aligner Purpose BLAST Many reference genome BLAT Large gaps (introns) BWA Small gaps (indels) Exonerate Ease of use GMAP Large gaps (introns) MUMmer Align two genome
- 72. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Three major challenges • Short reads (36-125 nt) • Error rates are considerable • Many reads span exon-exon junctions Alignment should be conducted on • Genome • Reference transcriptome Short read aligners are • No gaps allowed, or • Allow small gaps • Nether will work on intron regions RNA - seq alignment
- 73. Computational Biology and Genomics Facility, Indian Veterinary Research Institute RNA Exon 1 Exon 2 Exon read mapping Spliced read mapping Exon 1 Exon 2 RNA Seed matching K-mer seeds Seed extend RNA Exon 1 Exon 2 Exon read mapping Spliced read mapping Exon 1 Exon 2 RNA Seed matching K-mer seeds Seed extend RNA -seq alignment strategy Exon - first approach Seed - extend approach
- 74. Computational Biology and Genomics Facility, Indian Veterinary Research Institute • Available tools: • MapSplice, SpliceMap, TopHat • Two step procedure • Map reads continuously using unspliced read aligners • Unmapped reads are split into shorter segments and aligned independently • Efficient when not too many reads into the junction • Second step is computationally intensive • Can miss reads across exon-intron junctions RNA Exon read mapping Spliced read mapping Exon 1 Exon 2 Exon - first approach
- 75. Computational Biology and Genomics Facility, Indian Veterinary Research Institute • Representative algorithms • Genomic short read nucleotide alignment program (GSNAP) • Computing accurate spliced alignments (QPALMA) • Steps • Break reads into short seeds • Candidate regions are combined’ (such as Smith-Waterman) • Increased sensitivity • One arm may not provide enough specificity for alignment RNA Exon 1 Exon 2 Exon read mapping Spliced read mapping Exon 1 Exon 2 RNA Seed matching K-mer seeds Seed extend Seed - extend approach
- 76. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Sequence Assembly
- 77. Computational Biology and Genomics Facility, Indian Veterinary Research Institute • Overlap, layout, consensus • De Bruijn Graph or k-mer • Burrows Wheeler transform and FM-Index Source (Genome, Exome, Clones and amplicons,Transcriptome) Assembly (Reference-based assembly, de novo assembly) Assembly Algorithms -
- 78. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Algorithm Purpose OLC Small genome Long reads Handles indels De Bruijin graph Large genome Short high-quality reads No indels BWT and Ferragina- Manzini index Large genome Short or long No indels (currently) Assembly algorithms
- 79. Computational Biology and Genomics Facility, Indian Veterinary Research Institute Algorithm Assemblers OLC ARACHNE, CAP3, Celera assembler,MIRA,Newbler,Phrap De Bruijin graph ABySS, ALLPATHS, SOAP de novo, Velvet BWT and Ferragina- Manzini index String Graph Assembler (SGA) Assemblers
- 80. Computational Biology and Genomics Facility, Indian Veterinary Research Institute • Find two sequences with the largest overlap and merge them; repeat • Flaw: prone to mis - assembly Greedy Overlap, Layout, Consensus • Overlap • Find all pairs of sequences that overlap • Layout • Remove redundant and weak overlaps. • Merge pairs of sequences that overlap • unambiguously; that is, pairs of sequences that overlap only with each other and no other sequence • Consensus • Call the consensus base at positions where reads overlap