Neonatal nasal obstruction final
•Transferir como PPTX, PDF•
2 gostaram•563 visualizações

Neonatal nasal obstruction

Recomendados
Mais conteúdo relacionado
Mais procurados
Mais procurados (20)
Semelhante a Neonatal nasal obstruction final
Semelhante a Neonatal nasal obstruction final (20)
Mais de Arul Lakshmanaperumal
Mais de Arul Lakshmanaperumal (20)
Último
Último (20)
Neonatal nasal obstruction final
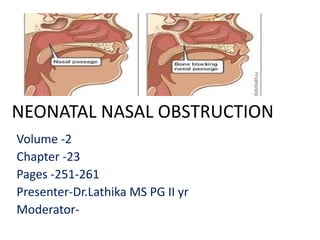
- 1. NEONATAL NASAL OBSTRUCTION Volume -2 Chapter -23 Pages -251-261 Presenter-Dr.Lathika MS PG II yr Moderator-
- 2. INTRODUCTION • The etiology of neonatal nasal obstruction is diverse. • Neonates are generally obligate nasal breathers for the first few months of life, and therefore they can present as acute respiratory emergencies, classically with cyclical cyanosis, relieved by crying. • The extent of their problems related to the neonate’s ability to breathe orally which is dependent on their maturity and neurological development.
- 3. INTRODUCTION • Thus, an oral airway is often sufficient to relieve the respiratory distress until definitive treatment can be undertaken. • Neonates with nasal obstruction may also present with stertor and feeding problems. Failure to thrive particularly raises level of concern.
- 4. Examination is essential! • Flexible nasal endoscopy is particularly useful • Imaging via computed tomography (CT) and magnetic resonance imaging (MRI) is of great value in delineating both nasal and post-nasal lesions.
- 6. CONGENITAL DISORDERS Skeletal anomalies CHOANAL ATRESIA • This is a rare condition (incidence 1 in 7000 live births)- complete obstruction of the posterior choanae on one or both sides • The blockage is either bony or membranous in origin, a mixed picture is usually seen (70% of cases), with the remainder being purely bony. • It is believed to be secondary to persistence of the nasobuccal membrane.
- 7. • Bilateral choanal atresia in neonates presents as acute respiratory distress as neonates are obligate nasal breathers. • Classically , cyclical cyanosis relieved by crying, and placement of an appropriatesized oral airway resolves the distress. • Unilateral choanal atresia may present later in life, and there is an inability to pass a nasogastric tube through one nasal passageway.
- 8. • Neonates with choanal atresia will have difficulty with feeding. • The uvula and epiglottis usually form a respiratory channel from the nose to the larynx with two lateral pathways from the mouth to the oesophagus to allow for the safe passage of food. • In neonates with bilateral choanal atresia this respiratory channel is lost and therefore cyanosis can develop during feeds.
- 9. • McGovern nipples have been shown to to be of benefit for children who develop feeding difficulties. • Misting upon placement of a metal spatula below the neonate’s external nasal aperture excludes a diagnosis of choanal atresia, and this test can easily be performed in the clinic setting.
- 10. • If suspected, the diagnosis should be confirmed with flexible nasendoscopy, and CT scanning should then be performed to determine the extent and nature of the choanal atresia (with suction clearance of the nose and application of 0.5% ephedrine drops 30 minutes prior to scanning. • In neonates, often a simple oral airway is well tolerated, in which case endotracheal intubation can be avoided.
- 14. CHARGE SYNDROME • CHARGE syndrome, due to mutations in the CHD7 gene on chromosome 8. • Therefore the minimum investigations in addition to the nasal CT scan are • cardiac echo, • renal ultrasound scan, and • ophthalmology • audiology review.
- 15. Surgical management- various approaches • The two most common techniques for choanal atresia repair are the transnasal and transpalatal approaches. • But the sublabial, transantral and transseptal approaches have also been described. • Transpalatal and transnasal surgery have been shown to have similar outcomes. • Transpalatal technique is not as common now, but it can be useful in craniofacial anomalies where the dimensions of the nose and postnasal space are limited. TRANSPALATAL APPROACH
- 16. • There are two methods described for the endoscopic transnasal approach. One involves using the zero degree endoscope transnasally, with serial dilatations using urethral sounds or using powered instruments such as microdrills. • In cases where the nasal cavity is too small to accommodate both instruments a posterior septal window is created and expanded, thus allowing the endoscope through one nostril and the powered instrument through the other nostril, creating a ‘neo-unichoana’. TRANSNASAL APPROACH
- 17. • The second transnasal approach involves a 120-degree endoscope being placed in the mouth and positioned in the nasopharynx behind the soft palate to give a view of the postnasal space. • Instruments and the drill can then be introduced through the nose. USING 120’ ENDOSCOPE
- 18. • There are reports in the literature of high success rates using the endoscopic endonasal approach with balloon dilatation for choanal atresia, although the numbers involved in these series are still quite small. • The role of nasal stenting post choanal atresia repair is also debated. If used, bilateral nasal stents can be fashioned from two ivory Portex™ endotracheal tubes cut to length with the bevelled end of each sitting in the nasopharynx orientated towards the septum. • The philtrum is protected by either a small length of size 12 suction catheter cut to act as a bridging piece or a further small piece of endotracheal tube. • The stents are secured by a circumseptal ‘0’ prolene suture and left in situ for up to 6 weeks Bilateral nasal stents with an endotracheal tube bridging piece.
- 19. REGULAR SUCTIONING! • A systematic review with metaanalysis has shown that the success rates for bilateral choanal atresia repair are similar with and without nasal stents, and that the use of stents may be associated with more complications • There is evidence that regular suctioning to clear secretions and daily washing with sodium chloride solution results in successful outcomes. • Authors who do not support using stents stress the need for resection of the posterior aspect of the vomer and early (1 week post repair) repeat examination for removal of granulations and dilatation as required.
- 20. Mitomycin C and KTP Laser • Is thought to reduce granulation tissue and fibrosis by inhibiting fibroblasts and angiogenesis leading to its use during stent removal. • However, several papers have found no benefit in terms of outcomes whether mitomycin c is used or not. • However, that mitomycin does have beneficial effects. • The ktp laser has also been shown to be helpful in the treatment of granulation tissue which develops post-operatively.
- 21. PIRIFORM APERTURE STENOSIS • This abnormality, first described in 1988, is a very rare condition leading to nasal obstruction in the neonate which arises due to bony overgrowth of the nasal process of the maxilla . • The piriform aperture is the narrowest part of the nasal airway and so even minimal reduction in diameter here can cause significant problems. • Symptoms similar to bilateral choanal atresia occur and epiphora is also often seen secondary to bony involvement of the nasolacrimal ducts. Diagnosis is suggested by the inability to pass a narrow gauge nasogastric tube or 2.2 mm endoscope through the anterior nasal vestibule due to the bony obstruction. CT scan (axial view) of bilateral piriform aperture stenosis
- 22. DIAGNOSIS • CT scan confirms the diagnosis with an aperture width of less than 11 mm measured on an axial CT at the level of the inferior meatus (in a term neonate). CT can also demonstrate a single central incisor, which exists in some affected individuals. • This single central incisor is associated with an absent upper frenulum and arch-shaped lower lip. In this subgroup with a ‘megaincisor’ there is a suggested association with holoprosencepaly, a rare condition in which the developing forebrain fails to divide appropriately to form the cerebral hemispheres, diencephalon, and optic and olfactory bulbs. • These patients should undergo further evaluation for central nervous system defects with an MRI and particularly the hypothalamic–pituitary–thyroid axis.
- 23. Note the single central incisor. MEGAINCISOR
- 24. • There are variable reports on the incidence rates of this condition with piriform aperture stenosis, but a figure of around 50% is generally accepted. • Conservative treatment with nasal steroid drops or decongestants (for up to 2 weeks) and saline irrigation is generally recommended as first-line treatment. • If there is severe obstruction, respiratory distress or failure to thrive, surgical treatment is warranted. • It has also been found that an aperture of less than 5 mm on CT is almost always associated with the need for surgical intervention. • Bilateral nasal stents with an endotracheal tube bridging piece.
- 25. SURGICAL MANAGEMENT • Surgery involves either a transnasal approach with an alar- releasing incision or • a sublabial approach with a gingival–buccal sulcus incision and elevation of the soft tissue and periosteum to expose the piriform aperture. • The abnormal bone is drilled away using a diamond burr and the mucoperiosteal flap replaced.
- 26. POST OP -FOLLOWUP • Post-operatively nasal stents can be used for up to 4 weeks, although more recent studies suggest that stenting is not necessary. • Complications include Adhesions, Septal perforations and Septal ulceration. • The use of suctioning, nasal irrigation and treating gastro-oesophageal reflux minimizes this
- 27. MIDNASAL STENOSIS • Midnasal stenosis is a rare condition secondary to overgrowth of the nasal bones halfway along the nasal cavity. • It usually occurs in association with syndromes characterized by midfacial hypoplasia, such as Apert syndrome, but cases in isolation are also reported. • Neonates will present in a similar fashion to those with piriform aperture stenosis or choanal atresia with apnoea, cyanosis and failure to thrive. • Diagnosis can be confirmed with nasal endoscopy or CT scanning which will demonstrate isolated bony narrowing of the midpart of the nasal cavity or narrowing with stenosis of the rest of the nasal cavity • Treatment is usually conservative, allowing the child’s midface to grow, such that by the age of 6 months the obstruction is relieved. • • For those children struggling with significant respiratory problems or failure to thrive, dilatations or stent placement can be considered.
- 28. Midnasal stenosis associated with a patient with Apert syndrome
- 29. NASAL AGENESIS • Complete arhinia is very rare but can occur in isolation or as part of a syndrome. • It originates at the fifth week in utero when the nasal placode fails to canalize to form the nasal passages. • Presentation at birth with acute respiratory distress occurs. • Management is initially with an oral airway and tube feeding. • A tracheostomy may be required. • Definitive surgical treatment usually involves a two-staged procedure aimed at reconstructing the nasal cavity as well as the external nose, and is usually delayed until facial development is almost complete.
- 30. Congenital nasal cysts DERMOID CYST • Dermoid cysts arise from the ectoderm and mesoderm and usually contain all the structures of normal skin. • They are the most common midline nasal mass, and account for between 1% and 3% of all dermoids. • Occasionally these dermoids can become infected and thus present as an abscess requiring drainage. • Between 4% and 45% of dermoid cysts have an intracranial component, thus pre-operative imaging with CT (for bony anatomy) and MRI (to delineate any connection to the central nervous system) is essential.
- 31. DERMOID CYST • These cysts usually present as a slowly growing cystic midline mass over the nasal dorsum. An associated pit is often seen in any position from the nasal tip to the glabella, and hair may be present at its opening.
- 32. NASOLACRIMAL DUCT CYST (DACRYOCYSTOCOELE) • The nasolacrimal duct system should canalize in utero from a superior to inferior direction and is usually complete by the sixth foetal month through a process of reabsorption; • however, not infrequently, at birth the lower end can remain closed. • This barrier can be combined with a proximal valve-like obstruction at the junction of the common canaliculus and lacrimal sac, thus the tear fluid builds up resulting in a cyst. • This is a common problem for neonates and it is reported that 5–30% of babies are born with nasolacrimal duct blockage.
- 33. DACRYOCYSTOCOELE • These lesions can cause epiphora and nasal obstruction, sometimes leading to respiratory distress and feeding difficulties, and may present with a bluish cystic mass at the medial canthus. • They are more commonly unilateral but can be bilateral, and their incidence is slightly higher in female infants. • CT imaging confirms the diagnosis and shows a dilated nasolacrimal duct, an intranasal cyst and cystic dilatation of the lacrimal sac. • Initial management is with nasal decongestants but, if surgical removal is required, endonasal marsupialization under endoscopic guidance is recommended. • Endonasal ablation with the carbon dioxide laser has also been • reported previously. • Opthalmology input is helpful as intra-operative nasolacrimal probing and stenting may be necessary.
- 34. THORNWALDT CYST • The pharyngeal recess or bursa sits in the midline of the posterior wall of the nasopharynx. • It ends next to the adenoids and is lined by the pharyngeal mucous membrane. • Cystic transformation of this recess was first described by Thornwaldt in 1885 and so it bears his name. • Inflammation of the lesion causes nasal obstruction, occipital pain, fullness in the ears and discharge. • It rarely causes significant obstruction in neonates. • Endoscopic examination confirms the diagnosis. • Imaging by CT and MRI demonstrates any adhesion to the cervical vertebrae. • Incision and excision of the cyst have been described while total clearance requires a palatal approach
- 35. NASOALVEOLAR CYSTS • These are rare, non-odontogenic, soft-tissue lesions arising from the incisive canal during the development of the maxilla. • They present lateral to the midline at the alar base and can cause asymmetrical alar flare. • Excision is usually via a sub-labial approach, but the transnasal approach has been recently reported. DENTIGEROUS CYSTS • Dentigerous cysts present in the floor of the nose or Maxillary sinus and have a dental origin. Endoscopic marsupialization or removal via the nose is usually satisfactory. MUCOUS CYSTS • Mucous cysts have been described anywhere in the nose but appear to be more common in the floor. • They may be congenital but are more usually seen as a complication of rhinoplasty. • Endoscopic and open approaches are used depending on the position of the lesion.
- 36. Nasal masses ENCEPHALOCOELE, MENINGOCOELE, GLIOMA • A nasal encephalomeningocoele represents a herniation of meninges with or without associated brain through bony defects of the calvarium. • A meningocoele consists of either meninges alone or with CSF and an encephalocoele contains nervous tissue. • Their combined incidence is around 1 in 4000 live births and they have an equal male/female distribution. • Encephalocoeles can be described as frontoethmoidal or basal. • Frontoethmoidal are usually associated with craniofacial deformity as they arise either at or anterior to the foramen caecum. • The basal types present intranasally through defects in the skull base causing nasal obstruction and widening of the nasal bridge.
- 38. Nasal gliomas • Benign midline masses containing glial cells and fibrous and vascular tissue. • They are similar to encephalocoeles but have become separated from the intracranial structures. Around 15% do, however, remain attached to the brain via a fibrous stalk. • There is usually no associated abnormality of the brain. • A better term for these lesions is ‘glial heterotopia’: glioma implies a neoplasm and these lesions are actually choristomas (aggregations of structurally normal tissue in an abnormal location). • Presentation is usually early on as a firm, non-compressible, reddish swelling.
- 40. • Differentiation between gliomas and encephalocoeles can be made in a number of ways. • A probe will pass laterally but not medially to an intranasal encephalocoele while an intranasal glioma can arise from the lateral nasalwall. • Furstenberg’s test (compression of the internal jugular vein) usually causes an encephalocoele to enlarge but a glioma does not. • Imaging is mandatory to confirm the nature of the lesion. • MRI is the most effective modality due to its better resolution of soft tissue, and because the anterior skull base contains unossified cartilage which can be mistaken for bony dehiscence on CT, but CT has a role in image guidance. • On MRI, an encephaocoele is seen as a mass in continuity with the brain with an associated skull base defect, while a glioma is discontinuous to the brain parenchyma and the tissue is dysplastic and gliotic therefore more hyperintense on T2 compared to normal brain parenchyma.
- 41. • Surgical excision is recommended for these masses, particularly if they are causing significant problems. • As with dermoid cysts, masses in the lower part of the nose can be removed via the external rhinoplasty approach. • More recently, the endoscopic approach is advocated. • The glial tissue can be removed with a zero- degree or 120- degree endoscope. • Encephalocoeles and meningocoeles that require surgery usually require a combined transnasal and neurosurgical approach. • Ventriculoperitoneal shunting may be required pre- operatively. • The intracranial portion can be excised via a bicoronal flap with a frontal craniotomy, but this can be associated with complications of epilepsy, anosmia, scarring and intracerebral haemorrhage.
- 42. • More recently, the endoscopic approach is advocated without the need for formal craniotomy. • The defect left by endoscopic excision can be closed with temporalis fascia graft, mucosa or a composite graft from the inferior turbinate, with Gelfoam®and packing (if small), or if a larger defect is present fascia lata and bone from the septum may be required. • This prevents the risk of CSF leak potentially leading to meningitis. • The role of prophylactic antibiotics is controversial.
- 44. NASAL HAEMANGIOMA • Vascular anomalies such as haemangiomas, arteriovenous malformations (AVMs) or vascular malformation (including lymphatic malformations) can present in the nose either externally or internally. • Internal haemangiomas often arise from the inferior turbinate. • Classically, a haemangioma is either absent or flat at birth and then undergoes a period of rapid growth to present as a mass at around 6 weeks of age. • Growth then continues for the first 6 months of life before gradual involution occurs, and the lesion generally disappears by around the age of 6 years. • This natural history supports conservative management if possible. • Ultrasound and MRI imaging are the recommended modes of imaging, particularly to exclude any intracranial connection, and treatment depends on the extent of involvement of the surrounding tissues.
- 45. TREATMENT • Treatment for haemangiomas has been transformed with the use of oral propranolol. • In cases where there is encroachment on the orbit with a potential risk to vision, surgical excision has been used to good effect. • The use of chemotherapy (such as methotrexate or vincristine) is reported but should be undertaken with caution due to the risks of side effects, and it has now largely been superseded by propranolol.
- 46. TERATOMA • A teratoma is a true neoplasm consisting of all three germ cell layers with cells varying in maturity. They occur in 1 in 4000 live births with less than 10% occurring in the head and neck. • The cervical forms are the most common, followed by nasopharyngeal teratomas. They are associated with polyhydramnios, stillbirth and prematurity, and can result in significant airway compromise. • They usually present as a firm mass. • Maternal serum alpha fetoprotein levels and beta HCG levels may be raised. • Imaging is with CT and MRI. • Teratomas will appear as heterogeneous masses on MRI, with fatty and bony components, and they may have a stalk, giving them mobility in different positions. • Management is surgical, either endoscopic or open, depending on the size of the lesion.
- 47. MISCELLANEOUS • Hamartomas, chordomas and craniopharyngiomas are extremely rare causes of nasal obstruction in the neonate Chordoma Chondromesenchymal hamartoma
- 48. ACQUIRED PATHOLOGIES 1 • Osseocartilaginous septal deformity 2 • Neonatal rhinitis 3 • Fibrous dysplasia 4 • Neoplasms of the nasal bones- Juvenile ossifying fibroma (JOF)
- 49. Osseocartilaginous septal deformity • The septum develops as an outgrowth from the merged medial nasal processes and nasofrontal process. • At week 9, it fuses with the palate just posterior to the incisive foramen, and then fuses anteriorly and posteriorly. • A number of babies are born with a septal deviation either in isolation or in association with an abnormality of the bony pyramid. • It is felt that the problem is due either to intrauterine positioning or to birth trauma.
- 50. Osseocartilaginous septal deformity • Closed reduction of the septal deformity with topical anaesthetic in each nostril in the first few days of life has been described and is thought to be successful if the deviation is severe. • However, most of the studies that advocate intervention have inadequate follow-up periods and there is little evidence for the adverse effects of conservative management. • Formal surgical repair is generally recommended later in childhood to avoid damage to the main growth centre of the nose; the external rhinoplasty approach has been used for other pathology in very young children and no detrimental effects on nasal growth have been reported
- 51. Neonatal rhinitis • Swelling of the nasal mucosa in newborn infants can cause significant airway problems, particularly when feeding, as neonates are obligate nasal breathers. • Idiopathic neonatal rhinitis is characterized by mucoid rhinorrhoea with nasal mucosal oedema in the afebrile newborn. • This results in stertor, poor feeding and respiratory distress. • Structural abnormalities should be excluded. • Treatment of neonatal rhinitis depends on the severity of symptoms. • Nasal bulb suction with saline drops in the first instance is recommended. • A short course of nasal steroid drops would be the next step. This should be closely monitored to avoid the potential side effects from systemic absorption
- 52. Neonatal rhinitis • It is important to consider chlamydia infection acquired in the birth canal. • This usually results in conjunctivitis but involvement of the nose is seen in around 25% of affected individuals. • Presentation is with obstruction, rhinorrhoea and a markedly erythematous nasal mucosa on examination. • Swabs are diagnostic and the appropriate antibiotics should be given. • Rarely congenital syphilis (Treponema pallidum) can cause nasal symptoms in the neonate. • Thin, clear secretions are seen between the second week and third month of life. This progresses to a mucopurulent discharge with significant obstruction and crusting of the nostrils. • Antibiotic treatment is required both for symptomatic relief and to prevent chronic infection of the cartilage resulting in saddle deformity.
- 53. Fibrous dysplasia • This is an uncommon cause of nasal obstruction in older children and young adults. • It is a benign fibro-osseous dysplasia and can present either as a solitary lesion (monostotic) or less commonly in multiple sites (polyostotic), typically in the craniofacial bones. • Presentation is usually as pain with progressive facial deformity between the ages of 10 and 30. • Nasal obstruction, with a mass on endoscopy or facial deformity due to growth of a lesion in the nose or sinus should raise suspicion. • Imaging helps to confirm the diagnosis; normal healthy bone is replaced with a more radiolucent ‘ground-glass’ appearance. • There can be endosteal scalloping of the inner cortex with a smooth non-reactive periosteal surface. • Lesions have diffuse margins.
- 54. Fibrous dysplasia • Management is expectant but surgical excision may be needed with the aim of preserving function and limiting disability. • The mid-facial degloving approach has been shown to achieve good results with minimal cosmetic defect. Craniofacial polyostotic fibrous dysplasia
- 55. Fibrous dysplasia • Medical treatment involves medication to increase bone density, for example biphosphonates. • A subgroup of polyostotic patients (around 3%) have • Associated endocrine abnormalities such as hyperthyroidism, adrenal disorders, diabetes, hyperpituitarism and hypercalcaemia with cafe-au- lait spots. • This is termed McCune–Albright syndrome after the two physicians who first described it in 1937. • There is a 1% risk of malignant transformation, mostly in the polyostotic form.
- 56. Neoplasms of the nasal bones • Juvenile ossifying fibroma (JOF) is a true neoplasm which is defined radiologically as a radiolucent, expansile, welldefined lesion with variable calcification. • It can be unilocular or multilocular with cortical thinning and possible perforation. Pain is rare. • There are two subtypes, trabecular and psammomatoid, which have different histopathological appearances. • Surgical excision is recommended and this may need to radical as recurrence rates are high (30–50%) probably due to the propensity of this disease to perforate cortical bone. • Malignant change has not been reported.