Chediak higashi syndrome, Phenylketonuria, Dentinogenesis Imperfecta
•Download as PPT, PDF•
33 likes•17,427 views


Recommended
More Related Content
What's hot
What's hot (20)
Viewers also liked
Viewers also liked (20)
Similar to Chediak higashi syndrome, Phenylketonuria, Dentinogenesis Imperfecta
Similar to Chediak higashi syndrome, Phenylketonuria, Dentinogenesis Imperfecta (20)
More from Janine Rumbaoa
More from Janine Rumbaoa (13)
Recently uploaded
Recently uploaded (20)
Chediak higashi syndrome, Phenylketonuria, Dentinogenesis Imperfecta
- 1. CHEDIAK HIGASHI SYNDROME
- 2. Chédiak–Higashi syndrome is a rare autosomal recessive disorder that arises from amicrotubule polymerization defect which leads to a decrease in phagocytosis. The decrease in phagocytosis results in recurrent pyogenic infections, partial albinism and peripheral neuropathy Mutations have been found in the CHS1 (also called LYST) gene. The primary defect in this disease is in special granules present in skin pigment cells and certain white blood cells
- 3. Chédiak–Higashi syndrome is caused by mutations in the LYST gene. This gene provides instructions for making a protein known as the lysosomal trafficking regulator
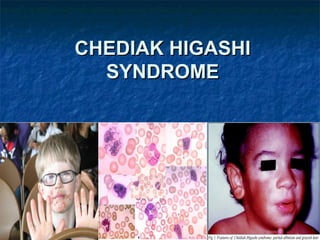
- 4. SIGNS AND SYMPTOMS Light skin Silvery hair Frequently complain of solar sensitivity Photophobia Infections involve mucous membranes, skin, and respiratory tract. Neuropathy are common
- 6. ACCELERATED PHASE Most children with Chédiak–Higashi syndrome ultimately reach a stage known as the accelerated phase — the lymphoma-like-syndrome. This severe phase of the disease is thought to be triggered by a viral infection (usually the Epstein–Barr virus, EBV). In the accelerated phase, defective white blood cells divide uncontrollably and invade many of the body's organs. The accelerated phase is associated with fever, episodes of abnormal bleeding, overwhelming infections, and organ failure. These medical problems are usually life-threatening in childhood.
- 7. Diagnosis The diagnosis is confirmed by bone marrow smears that show "giant inclusion bodies" in the cells that develop into white blood cells (leukocyte precursor cells). CHS can be diagnosed prenatally by examining a sample of hair from a fetal scalp biopsy or testing leukocytes from a fetal blood sample
- 8. Under light microscopy the hairs present evenly distributed, regular melanin granules, larger than those found in normal hairs. Under polarized light microscopy these hairs exhibit a bright and polychromatic refringence pattern
- 10. TREATMENT There is no specific treatment for Chédiak– Higashi syndrome. Bone marrow transplants appear to have been successful in several patients. Infections are treated with antibiotics and abscesses are surgically drained when appropriate. Antiviral drugs such as acyclovir have been tried during the terminal phase of the disease. Cyclophosphamide and prednisone have been tried. Vitamin C therapy has improved immune function and clotting in some patients
- 11. PHENYLKETENURIA
- 12. Phenylketonuria (commonly known as PKU) is an inherited disorder that increases the levels of a substance called phenylalanine in the blood. Phenylalanine is a building block of proteins (an amino acid) that is obtained through the diet. It is found in all proteins and in some artificial sweeteners. If PKU is not treated, phenylalanine can build up to harmful levels in the body, causing intellectual disability and other serious health problems.
- 13. SIGNS AND SYMPTOMS Infants with classic PKU appear normal until they are a few months old. Without treatment with a special low-phenylalanine diet, these children develop permanent intellectual disability. Seizures, delayed development, behavioral problems, and psychiatric disorders. Untreated individuals may have a musty or mouse-like odor as a side effect of excess phenylalanine in the body. Children with classic PKU tend to have lighter skin and hair than unaffected family members and are also likely to have skin disorders such as eczema.
- 14. Less severe forms of this condition, sometimes called variant PKU and non- PKU hyperphenylalaninemia, have a smaller risk of brain damage. People with very mild cases may not require treatment with a low- phenylalanine diet. These infants may also have a low birth weight and grow more slowly than other children. Other characteristic medical problems include heart defects or other heart problems, an abnormally small head size (microcephaly), and behavioral problems. Women with PKU and uncontrolled phenylalanine levels also have an increased risk of pregnancy loss.
- 15. (A), trace diffusion-weighted (B), and ADCav (C) images show extensive white matter abnormalities with restricted diffusion.
- 16. DIAGNOSIS PKU can be easily detected with a simple blood test. All states in the US require a PKU screening test for all newborns as part of the newborn screening panel. The test is generally done by taking a few drops of blood from the baby before the baby leaves the hospital. If the initial screening test is positive, further blood and urine tests are required to confirm the diagnosis.
- 17. TREATMENT Treatment involves a diet that is extremely low in phenylalanine, particularly when the child is growing. The diet must be strictly followed. This requires close supervision by a registered dietitian or doctor, and cooperation of the parent and child. Those who continue the diet into adulthood have better physical and mental health. “Diet for life” has become the standard recommended by most experts. This is especially important before conception and throughout pregnancy. A special infant formula called Lofenalac is made for infants with PKU. It can be used throughout life as a protein source that is extremely low in phenylalanine and balanced for the remaining essential amino acids
- 19. Dentinogenesis imperfecta (hereditary Opalescent Dentin) is a genetic disorder of tooth development. causes teeth to be discolored (most often a blue- gray or yellow-brown color) and translucent. Teeth are also weaker than normal, making them prone to rapid wear, breakage, and loss. These problems can affect both primary (baby) teeth and permanent teeth. This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.
- 20. TYPES TYPE I - usually an autosomal dominant trait with variable expressivity but can be recessive if the associated osteogenesis imperfecta is of recessive type - usually involved and more severely affected are deciduous teeth in type 1
- 21. TYPE II - Occurs in people without other inherited disorders (i.e. Osteogenesis imperfecta).It is an autosomal dominant trait. A few families with type II have progressive hearing loss in addition to dental abnormalities.
- 22. CLINICAL FEATURES The teeth may be gray to yellowish brown. They exhibit translucent or opalescent hue. Enamel is usually lost early due to loss of scalloping at the DEJ. However, the teeth are not more susceptible to dental caries than normal ones.
- 23. RADIOGRAPGIC APPEARANCE Type I and II show total obliteration of the pulp chamber. Type III shows thin dentin and extremely enormous pulp chamber.These teeth are usually known as Shell Teeth . Histology Dentinal tubules are irregular and are bigger in diameter. Areas of uncalcified matrix are seen. Sometimes odontoblasts are seen in dentin .
- 25. TREATMENT One treatment option is bonding, putting lighter enamel on the weakened enamel of the teeth and with lots of treatments of this bonding, the teeth appear whiter to the eye, but the teeth on the inside and under that cover are still the same. Due to the weakened condition of the teeth, many common cosmetic procedures such as braces and bridges are inappropriate for patients with Dentinogenesis imperfecta and are likely to cause even more damage than the situation they were intended to correct.