Reduced representation bisulfite sequencing identified differential hypermethylation of the c-MER proto-oncogene (MERTK) in approximately 25% of colon cancer cell lines and tumors. Rapid amplification of cDNA ends showed predominantly 5' truncated MERTK mRNA transcripts in methylated colon cancer cell lines. The document aims to determine the mechanism by which hypermethylation alters MERTK expression and transcript structure. The authors hypothesize that methylation causes alternative splicing producing a constitutively active truncated tyrosine kinase. They plan to clone truncated cDNA fragments into cell lines to assess effects on MERTK activation, cell growth, and subcellular localization.
1. Determining Effect of DNA Hypermethylation of c-mer proto-oncogene (MERTK)
on Gene Expression and Transcript Structure
DNA methylation is identified as an essential epigenetic modification regulating gene
expression in normal development and has critical roles in genomic imprinting and X-
chromosome inactivation. However, aberrant methylation of CpG islands in DNA promoters has
also been established as an abnormal finding in human colon cancers and can be a
mechanism to silence or alter gene expression.
We employed reduced representation bisulfite sequencing (RRBS) which utilizes restriction
enzyme digest and bisulfite conversion of genomic DNA on a reduced fraction of the genome
with high CpG content to identify areas of differential methylation between normal and
neoplastic colon tissue. Through this comparison, we identified c-MER proto-oncogene
(MERTK) as differentially hypermethylated in a subset (~25%) of colon cancer cell lines and
both primary and metastatic tumors. This proto-oncogene expresses a receptor tyrosine kinase
that is often overexpressed or activated in various malignancies including melanoma.
In order to determine the molecular mechanism by which hypermethylation results in altered
expression of MERTK, we performed rapid amplification of cDNA ends (RACE) in
unmethylated and methylated colon cancer cell lines and found predominantly 5’ truncated
mRNA transcripts in the methylated cell lines, but not in the unmethylated. The truncated
transcripts suggest alternative splicing mechanisms that could result in a constitutively active
“rouge” tyrosine kinase. To further assess the significance of the truncated transcripts, we
attempt to clone complementary DNA fragments of the truncated mRNA into plasmid vectors
that will be transformed into and expressed by mammalian cell lines. Finally, we evaluate if
expression of truncated kinase results in constitutive activation of MERTK, changes in cell
growth (proliferation), and incorporate immunofluorescence techniques to assess different
subcellular localization of the receptor tyrosine kinase.
Presenter: David Dornblaser
Collaborators: Ryan Fecteau, Helen Moinova, Sanford Markowitz
Case Comprehensive Cancer Center, Case Western Reserve University, Cleveland, OH
Abstract
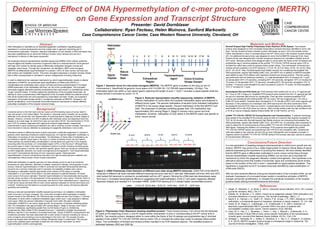
Figure 1. Genomic locus of c-mer proto-oncogene (MERTK). The MERTK gene is located on the long (q) arm of
chromosome 2. Specifically its genomic locus spans chr2:112,656,191-112,786,945 (approximately 130 kbp). The
methylated region lies within a CpG island (green) spanning the length of exon 1. Exon 1 encodes a signal peptide while the
kinase domain is encoded by exons 12-19.
Roche® Expand High Fidelity Polymerase Chain Reaction (PCR) System: Two forward
primers were designed to mirror truncated transcription products that were identified in tumor cell
lines. The two forward primers included a primer that began at exon 2 (E2) and another primer
further downstream in exon 2 corresponding to the 67th amino acid in the receptor tyrosine kinase
(67). An additional forward primer was designed to mirror the full MERTK transcript (Full). Each
forward primer was designed with a Kozak consensus sequence for translation initiation and an
ATG codon. Reverse primers were designed either to clone within the frame of the V5 epitope and
polyhistidine tag (C-terminal peptide) of the pcDNA TM3.1/V5-His TOPO® cloning vector (V5) or
included the native stop codon to express native protein (Stop). The combination of forward and
reverse primers resulted in six PCR reactions: (1) Full, V5; (2) Full, Stop; (3) E2, V5; (4) E2, Stop;
(5) 67, V5; (6) 67, Stop. Roche® expand high fidelity enzyme mix containing thermostable Taq
DNA polymerase, expand high fidelity buffer (10x), and dNTP mix in addition to MERTK template
were added to each PCR reaction with respective forward and reverse primers. Thermal cycling
was performed with the following conditions: (1) initial denaturation 94oC (5 minutes) for 1 cycle;
(2) denaturation 94oC (15 seconds), annealing 62oC (30 seconds), elongation 72oC (2 minutes) for
10 cycles; (3) denaturation 94oC (15 seconds), annealing 62oC (30 seconds), elongation 72oC (2
minutes + 5 second cycle elongation for each successive cycle) for 15 cycles; (4) final elongation
72oC (7 minutes) for 1 cycle.
NucleoSpin® Gel and PCR Clean-up: PCR products were loaded and run on a 1% agarose gel
for 1 hour at 150 V (Figure 4). Amplified PCR products were excised from the 1% agarose gel with
the aid of a ultraviolet light source for visualization. Each gel slice containing amplified PCR
product was solubilized with binding Buffer NTI (for every 100 mg of agarose gel < 2% 200 µl
buffer NTI were added). Samples were incubated for 5-10 minutes at 50oC and vortex agarose gel
dissolved. In the presence of a chaotropic salt, DNA was bound to the silica membrane of the
NuceloSpin® Gel and PCR Clean-up Column. Contaminations were removed by adding ethanolic
wash Buffer NT3 to the PCR Clean-up Column. DNA is finally eluted under low salt conditions into
a 15-30 µl volume of slightly alkaline Elution Buffer NE (5 mM Tris/HCl, pH 8.5).
pcDNA TM3.1/V5-His TOPO® TA Cloning Reaction and Transformation: 3’ adenine overhangs
were added to the amplified PCR products using an Add an A protocol that required incubation at
72oC for 20 minutes with Roche® expand high fidelity enzyme mix containing thermostable Taq
DNA polymerase, expand high fidelity buffer (10x), and dATP mix. A-overhangs were necessary in
order to utilize the TA cloning scheme of the pcDNA TM3.1/V5-His TOPO® that does not requires
DNA ligase. Following the addition of A-overhangs, PCR products were ligated into pcDNA
TM3.1/V5-His TOPO® vectors and transformed into TOP10 E.coli competent cells. Transformed
cells were plated in two volumes (20 and 200 µl) onto LB/Ampicillin and incubated overnight. A
negative control of pcDNA TM3.1/V5-His TOPO® without PCR product was also cultured to assess
for transformation efficiency.
Materials and Methods
Figure 4. Polymerase Chain Reactions showing amplified product. Three forward primers: Full mirrors the full transcript,
E2 starts at the beginning of exon 2, and 67 begins farther downstream in exon 2 corresponding to the 67th amino acid in
MERTK. Two reverse primers: designed either to clone within the frame of the V5 epitope and polyhistidine tag (C-terminal
peptide) of the pcDNA TM3.1/V5-His TOPO® cloning vector (V5) or included the native stop codon to express native protein
(Stop). The combination of forward and reverse primers resulted in six PCR reactions (above). The amplified product is
estimated between 2500 and 3000 bp.
Colorectal cancer is the third most common cancer and third leading cause of cancer death in
men and women in the United States. The decline in colorectal cancer mortality can be partially
attributed to the introduction and dissemination of screening tests to diagnose at earlier stages of
disease. However, currently only 40% of patients with colorectal cancer are diagnosed when the
disease is at a local stage, for which the 5-year survival rate is 90.3%. Survival declines to 70.4%
and 12.5% for patients diagnosed with regional and distant-stage disease, respectively1.
Improvements in early screening and detection are vital to continue to lower mortality. One area of
focus has been to improve detection by assessing for epigenetic alterations in tumor cells.
Inheritance based on differential levels of gene expression constitutes epigenetics in contrast to
genetics which describes inheritance of gene sequences. An important epigenetic modification is
methylation of cytosines at CpG dinucleotides. The distribution of these sites of methylation can
be localized to specific tissue types in the body in a series of patches known as CpG islands.
These patches are predominantly unmethylated in normal tissue and span the 5’ end of genes
traversing either the promoter, a 5’ untranslated region (UTR), or the first exon2. Although there
are several cases in which CpG island methylation functions normally including imprinted genes,
X-chromosome genes in women, and germline-specific genes, there are numerous mechanisms
by which epigenetic alteration can cause tumorigenesis and progression3. CpG hypermethylation
can result in transcriptional silencing of tumor suppressor genes. Global genomic
hypomethylation of cancer cell genomes may also occur that promotes genomic instability and
carcinogenesis without proper control of gene expression2.
Differential methylation of specific genomic loci have already proven to serve as functional
biomarkers for early detection, detection of relapse, response to therapy, and prognosis of colon
cancers. Aberrant hypermethylation of hMLH1 promoter and resultant transcriptional silencing
proved to be a common molecular event in sporadic microsatellite unstable colon cancer.
Developing a methylation-specific polymerase chain reaction (PCR) assay to evaluate
methylation in a CpG island of the hMLH1 promoter allowed for potential detection of human
colon cancers from serum samples.4 Aberrant hypermethylation of transcriptionally silent genes
has also shown promise in the development of new screening for colon cancer. Vimentin, which is
transcriptionally inactive in normal colonocytes, was found to have methylated exon-1 sequences
in both tumor tissue and fecal DNA of colon cancer patients compared to normal colon tissue and
controls5. Including new biomarkers to a screening panel for colon cancer will improve detection
specificity and sensitivity.
Utilizing a reduced representation bisulfite sequencing technique, we validated a methylation
event occurring in a known oncogene, the c-mer proto-oncogene which expresses a receptor
tyrosine kinase of the TAM family. The TAM family also includes Axl and Tyro3 (Figure 1). Percent
methylation of CpGs within a localized methylated region within exon-1 was analyzed in different
tumor types. While the percent methylation of several CpGs within the patch indicated
methylation of MERTK is not cancer stage specific, the proportion of samples exhibiting greater
than 10% methylation was approximately 25% among Stage II, Stage IV primary colon cancer
and liver metastases, significantly greater than normal tissues (Figure 2). Additionally,
complementary DNA (cDNA) PCRs indicated that exon-1 methylation inhibited transcriptional
efficiency near the 5’ end resulting in truncated transcripts. (Figure 3) MERTK overexpression and
constitutive activation has been associated with a wide variety of cancers indicating its role as a
proto-oncogene and providing a survival advantage to the tumor cell. The receptor tyrosine
kinase has already been identified as a biologic therapeutic target in melanomas.6 We pursued
study of the validated gene candidate to determine the molecular mechanism by which
hypermethylation confers carcinogenesis.
Background
References
Conclusions/Future Direction
1. Siegel, R., DeSantis, C., & Jemal, A. (2014). Colorectal cancer statistics, 2014. CA: a cancer
journal for clinicians, 64(2), 104-117.
2. Esteller, M., & Herman, J. G. (2002). Cancer as an epigenetic disease: DNA methylation and
chromatin alterations in human tumours. The Journal of pathology, 196(1), 1-7.
3. Baylin S. B., Herman J. G., Graff J. R., Vertino, P. M., & Issa, J. P. (1997). Alterations in DNA
methylation: a fundamental aspect of neoplasia. Advances in cancer research, 72, 141–196.
4. Grady, W. M., Rajput, A., Lutterbaugh, J. D., & Markowitz, S. D. (2001). Detection of
aberrantly hypermethylated hMLH1 promoter DNA in the serum of patients with
microsatellite unstable colon cancer. Cancer research, 61(3), 900-902.
5. Chen, W. D., Han, Z. J., Skoletsky, J., Olson, J., Sah, J., Myeroff, L. & Markowitz, S. D.
(2005) Detection in fecal DNA of colon cancer-specific methylation of the nonexpressed
vimentin gene. Journal of the National Cancer Institute, 97(15), 1124-1132.
6. Schlegel, J., Sambade, M. J., Sather, S., Moschos, S. J., Tan, A. C., Winges A., & Graham,
D. K. (2013). MERTK receptor tyrosine kinase is a therapeutic target in melanoma. The
Journal of clinical investigation, 123(5), 2257.
Figure 2. Reduced representation bisulfite sequencing validation of MERTK.
Percent methylation of CpGs within the localized methylated region was analyzed in
different tumor types. The percent methylation of several CpGs indicated methylation
of MERTK is not cancer stage specific. Percent methlyation of the first MERTK CpG
(left). The proportion of samples exhibiting greater than 10% methylation was
approximately 25% among Stage II, Stage IV primary colon cancer and liver
metastases. However, methylation of CpG island in the MERTK patch was specific to
tumor versus normal.
In a new generation of targeting biological pharmaceuticals to inhibit tumor growth and cell
division, MERTK may prove to be a viable target protein to improve clinical status of cancer
patients emphasizing the importance of pursuing this research. We have already identified
that the MERTK gene is differentially methylated in tumor versus normal tissue through
reduced representation bisulfite sequencing (RRBS), but we have yet to elucidate the
mechanism by which the epigenetic alteration confers tumorigenesis. One hypothesis is an
alternative splicing event that creates a functionally rogue and constitutively active kinase
based on the location of the CpG in exon-1 responsible for signal peptide translation. This
hypothesis is supported by the 5’ truncated transcripts identified by rapid amplification of
cDNA ends (RACE).
After we have achieved effective cloning and transformation of the truncated cDNA, we will
evaluate if expression of a truncated kinase results in constitutive activation of MERTK,
changes cell growth (proliferation), or changes the subcellular localization of the receptor
tyrosine kinase utilizing immunofluorescence techniques.
Figure 3. cDNA Polymerase Chain Reaction at different exon sites along MERTK transcript. cDNA PCR of the MERTK
transcript in different cell lines indicated different transcript structure near the 5’ start site. Methylated cell lines v400 and v670
(red) lacked full transcript in contrast to unmethylated cell line v871 (green). Moving the cDNA start site downstream away
from exon-1 increased transcriptional efficiency suggesting that hypermethylation of the 5’ CpG patch negatively affected
transcript integrity and resulted in 5’ truncated mRNA. cDNA PCR starting at E13 yielded higher concentrated product.