

Recomendados
Mais conteúdo relacionado
Mais de Rodrigo Tinoco
Mais de Rodrigo Tinoco (20)
Metabolismo de lipídeos e proteínas
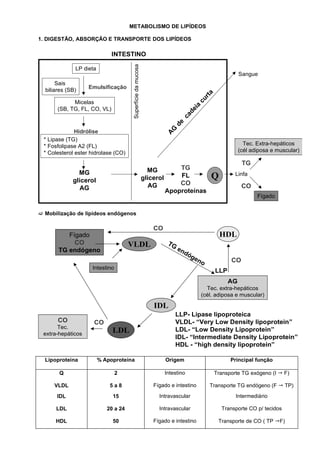
- 1. METABOLISMO DE LIPÍDEOS 1. DIGESTÃO, ABSORÇÃO E TRANSPORTE DOS LIPÍDEOS INTESTINO Superfície da mucosa LP dieta Sangue Sais Emulsificação biliares (SB) a rt cu Micelas a (SB, TG, FL, CO, VL) d ei ca de G Hidrólise A * Lipase (TG) * Fosfolipase A2 (FL) Tec. Extra-hepáticos * Colesterol ester hidrolase (CO) (cél adiposa e muscular) TG MG TG MG glicerol glicerol FL Q Linfa AG CO CO AG Apoproteínas Fígado Mobilização de lipídeos endógenos CO Fígado HDL CO VLDL TG TG endógeno en dó ge no CO Intestino LLP AG Tec. extra-hepáticos (cél. adiposa e muscular) IDL LLP- Lipase lipoproteica CO CO VLDL- “Very Low Density lipoprotein” Tec. extra-hepáticos LDL LDL- “Low Density Lipoprotein” IDL- “Intermediate Density Lipoprotein” HDL - “high density lipoprotein” Lipoproteina % Apoproteína Origem Principal função Q 2 Intestino Transporte TG exógeno (I F) VLDL 5a8 Fígado e intestino Transporte TG endógeno (F TP) IDL 15 Intravascular Intermediário LDL 20 a 24 Intravascular Transporte CO p/ tecidos HDL 50 Fígado e intestino Transporte de CO ( TP F)
- 2. Resumo
- 3. 2. DEGRADAÇÃO DOS TRIGLICERÍDEOS (LIPÓLISE) 2.1. Destino do glicerol; pode ser convertido em glicose (gliconeogênese), CO2 + H2O ( glicólise) ou triglicerídeos ( lipogênese); 2.2. Degradação oxidativa dos ácidos graxos – β - OXIDAÇÃO • Ocorre na matriz mitocondrial; • Pode ser dividida em 3 etapas: 2.2.1. Ativação dos AG: CH3-(CH2)n-COOH + ATP + CoA-SH CH3-(CH2)n-CO-S-CoA + AMP + PPi Acil-S-CoA 2.2.2. Transporte do Ácido graxo ativado (Acil-S-CoA)para o interior da mitocôndria Acil-S-CoA + carnitina Acil-carnitina + CoA-SH Acil-carnitina + CoA-SH Acil-S-CoA + carnitina 3. β -Oxidação do AG: Cada dois carbonos dos ácidos graxos irá gerar um AcetilCoA CH3-(CH2)8-CO-S-CoA + CoA-SH CH3-(CH2)6-CO-S-CoA + Acetil-CoA Acil-S-CoA A oxidação de ácidos graxos com mesmo número de carbonos insaturados é mais rápida do que a degradação dos ácidos graxos saturados;
- 4. A síntese do ATP vem da oxidação dos Acetil-CoA no cicli de Krebs e cadeia respiratória e do transporte de elétrons dos NADH e dos FADH2 na cadeia respiratória; 2.2.3. FORMAÇÃO DE CORPOS CETÔNICOS: • Ocorre no fígado; • Oxidação de AG Excesso de Acetil-CoA Oxidação ou convertido em corpos cetônicos (CC) - Acetoacetato, β -hidroxibutirato e Acetona; • CC são ácidos fortes (pKa ≅ 3,5) aumento de CC diminui pH do sangue impedindo a ligação da Hb ao O2; • Distribuição da energia obtida pela oxidação dos AG no fígado para todo o organismo tecidosperiféricos (mús. esquelético e coração) CC regeneram Acetil-CoA CK; 3. BIOSSÍNTESE DE TRIGLICERÍDEOS (LIPOGÊNESE) 1.1. Biossíntese de ácidos graxos • Ocorre no citosol da célula 1 ETAPA: Formação do malonil-CoA: Acetil-CoA (cit) + ATP + CO2 + H2O malonil-CoA + ADP + Pi + H+ 2 ETAPA: Síntese do ácido graxo (ácido palmítico): E: Sistema da çido graxo sintetase (enzima reguladora):
- 5. 3 ETAPA: Elongação e insaturação dos ácido graxos: • O ácido palmítico é precursor de outros AG de cadeia longa e de alguns AG insaturados • Elongação de AG: Ocorre no retículo endoplasmático e na mitocôndria através da adição de grupos acetil (malonil-CoA) • Insaturação cis ∆ 9 – reação oxidativa catalizada Acil-CoA oxigenase; Ác. palmítico (C16) dessaturação elongação Äc. Palmitoléico (C16 ∆9 ) Ác. esteárico (C18) Elongação dessaturação AG saturados superiores Ác. oleico (C18 ∆9 ) Plantas Linoléico (C18 ∆ 9,12 ) Plantas dessaturação -linolênico (C18 ∆ 9,12,15 ) -linolênico (C18 ∆6,9,12 ) elongação outros ácidos poliinsaturados dessaturação Araquidônico Prostaglandinas 3.2. Biossíntese de triglicerídeos: 1- AG + glicerol Triacilglicerol (TG) 4. REGULAÇÃO DO METABOLISMO DE ÁCIDOS GRAXOS Síntese: • Ativação: citrato e insulina; • Inibição: Acil-CoA, glucagon e epinefrina Degradação: • Ativação: glucagon ativa a enzima Lipase hormônio sensível (tecido adiposo) libera AG no sangue aumento da oxidação pelos músculos e fígado; • Inibição: Insulina, glicocorticóides, hormônio do crescimento inibem a enzima Lipase hormônio sensível (tecido adiposo) diminui a liberação de AG no sangue; 5.2. Degradação do colesterol O colesterol não pode ser metabolizado a CO2 e H2O; O colesterol é o precursor dos hormônios esteróides: glicocorticóides (ex: cortisol), mineralocorticóides ( ex: aldosterona) e hormônios sexuais ( andrógenos, estrógenos e progestógenos); Parte do colesterol sofre a ação de bactérias no intestino e é eliminado;
- 6. No fígado parte do colesterol é transformada em ácidos biliares primários sais biliares primários e secundários (fígado) canal biliar duodeno bactérias transformam os sais biliares novamente em ácidos biliares secundários parcialmente eliminados nas fezes (0,5g/dia); METABOLISMO DE PROTEÍNAS 1. DIGESTÃO: - Estômago: pepsinogênio (zimogênio – inativo) pepsina (enzima ativa) - Intestino Delgado: tripsinogênio, quimiotripsinogênio, procarboxipeptidase tripsina, quimiotripsina, carboxipeptidase, Aminopeptidases Proteínas Peptídeos Aminoácidos livres Capilares sanguíneos Fígado ver esquema do catabolismo de aminoácidos DEGRADAÇÃO OXIDATIVA DOS AMINOÁCIDOS • Aumentada: * Ingestão de excesso de aa com relação às necessidades p/ síntese proteica; * Jejum ou Diabetes mellitus – carboidratos não são disponíveis; * Aa liberados em excesso durante a degradação normal de proteínas; • Os esqueletos carbônicos dos aa são degradados por 20 vias diferentes • Todas as vias catabólicas dos aa convergem formando 5 produtos que entram no CK e são completamente degradados à CO2 e H2O: • Aminoácidos glicogênicos: - podem ser convertidos em piruvato ou intermediários do ciclo de Krebs • Aminoácidos cetogênicos – podem ser convertidos Acetil CoA Algumas doenças genéticas humanas que afetam o metabolismo de aa Nome Enzima ou processo defeituoso Aa envolvido Albinismo Tirosina 3-monoxigenase Tir Alcaptonúria Homogentisato 1,2-dioxigenase Fen Homocistinúria Cistationina -sintetase Cys Fenilcetonúria Fenilalanina 4- monoxigenase Fen Hipervalinemia Valina transaminase Val • Etapas da oxidação dos aminoácidos 1. Transferência dos grupos α-amino é catalisada pelas transaminases • Durante a degradação oxidativa ocorre a remoção dos grupos amino dos aa; • Se não utilizados pelo organismo estes grupos são coletados e convertidos em uréia e excretados; • Transaminação: α-amino é transferido do aa ao a-cetoglutarato formando o α-cetoácido correspondente e o glutamato: E: transaminases 2. A amônia é formada à partir do glutamato • Glutamato NH4+ A amônia é uma substância muito tóxica (neurotóxica), principalmente no cérebro (a amônia livre (NH3) pode penetrar as membranas e entrar nas células cerebrais e sua mitocôndria. A entrada de amônia na mitocondria diminui oxidação da glicose e a formação de ATP, diminui a produção de neurotransmissores e altera o pH celular. 3. A amônia se transforma em glutamina para ser transportada dos tecidos periféricos para o fígado • A glutamina é um composto não tóxico, neutro, que atravessa facilmente as membranas celulares; • Nos músculos é a alanina que transporta a amônia para o fígado (Ciclo da glicose-alanina) 2. No fígado a glutamina e alanina são convertidas novamente em amônia
- 7. 3. No fígado a amônia é convertida em uréia no ciclo da uréia • Defeitos genéticos no ciclo da uréia levam a um excesso de amônia no sangue (desordens mentais, desenvolvimento retardado podendo levar ao coma e morte. 4. A uréia é eliminada na urina ´ A EXCREÇÃO DO GRUPO AMINO PODE SER COMO: AMÔNIA, URÉIA OU ÁCIDO ÚRICO ANIMAIS AMONOTÉLICOS (peixes)- excreta amônia livre – ambiente aquático ANIMAIS UREOTÉLICOS (maioria dos animais terrestres) – excreta uréia (ciclo da uréia) ANIMAIS URICOTÉLICOS (pássaros, largatos e serpentes) – excreta ácido úrico- excreção do amino numa forma semi-sólida; 3. BIOSSÍNTESE DOS AMINOÁCIDOS NÃO ESSENCIAIS Aa essencias Aa não essenciais Arginina Alanina Histidina Asparagina Isoleucina Aspartato Leucina Cisteína Lisina Glutamato Metionina Glutamina Fenilalanina Glicina Treonina Prolina Triptofano Serina Valina Tirosina CONVERSÃO DOS AMINOÁCIDOS EM PRODUTOS ESPECIALIZADOS Os aminoácidos são unidades construtoras de proteínas e de muitos compostos nitrogenados com funções fisiológicas importantes, como por exemplo: • PORFIRINAS - heme (hemoglobina, mioglobina, citocromos, etc.) • CREATINA - Fosfocretina (depósito de fosfato de alta energia no múculo) . A degradação da creatina gera a creatinina que é excretada na urina • HISTAMINA – Sintetizada à partir da Histidina – poderoso vasodilatador reações alérgicas e inflamatórias; • SEROTONINA – Sintetizada à partir do Triptofano percepção de dor, regulação do sono, temperatura e pressão arterial; • CATECOLAMINAS - dopamina, norepinefrina e epinefrina. Sintetizadas à partir de Tirosina neurotransmissores (cérebro e sistema nervoso autônomo) e hormônios (controle do metabolismo de carboidratos e lipídeos) • MELANINA - Sintetizadas à partir de Tirosina pigmento que dá cor principalmente aos olhos, cabelos e pele.